负载型MoS2基加氢催化剂活性相的限域构建及其加氢脱硫活性
2024-06-24李彦鹏孙于林杨兆胜刘大鹏柴永明刘晨光
李彦鹏 孙于林 杨兆胜 刘大鹏 柴永明 刘晨光


摘要:基于对负载型NiMo加氢脱硫催化剂活性相的结构设计,以具有内径10~30 nm直通孔道的天然埃洛石纳米管(HNT)为载体,利用其“孔道空间限域效应”作为MoS2活性相结构的控制基础,以硫代钼酸铵和硝酸镍为金属前驱物,采用等体积浸渍法一次浸渍工艺,通过改变金属负载量、Ni/Mo(原子比)等工艺条件制备系列双金属NiMo/HNT催化剂。通过X射线衍射(XRD)、比表面积(BET)、高分辨透射电子显微镜(HRTEM-EDS mapping)、X射线光电子能谱仪(XPS)等手段对所得催化剂的结构进行表征,以二苯并噻吩(DBT)为模型化合物对催化剂的脱硫活性进行评价。结果表明:HNT载体的使用可有效控制MoS2活性相尺寸,催化剂中的Mo、Ni金属组分几乎全部集中于载体的孔道内部并呈原生的亲密接触状态,同时Ni、Mo组分具有更高的硫化程度,协同效果更好;NiMo/HNT催化剂的加氢脱硫活性和加氢选择性均显著优于使用氧化铝载体的参比催化剂,其中DBT转化率最高增幅41.6%,反应速率R最大可以提升92 %,加氢选择性最大可提升60%。
关键词:加氢脱硫; 活性相; 埃洛石纳米管; 孔道限域
中图分类号:TE 624.43 文献标志码:A
文章编号:1673-5005(2024)03-0207-08 doi:10.3969/j.issn.1673-5005.2024.03.023
Active-phase construction and hydrodesulfurization activity evaluation of supported MoS2-based hydrogenation catalysts designed with spatial confinement strategy
LI Yanpeng1,2, SUN Yulin1, YANG Zhaosheng1, LIU Dapeng1, CHAI Yongming1, LIU Chenguang1
(1.State Key Laboratory of Heavy Oil Processing in China University of Petroleum(East China), Qingdao 266580, China;2.Advanced Chemical Engineering and Energy Materials Research Center in China University of Petroleum(East China), Qingdao 266580, China)
Abstract:Based on the structural design of the active phase of the supported NiMo hydrodesulfurization catalyst, a “spatial confinement strategy” was adopted in order to achieve the structure design of the active MoS2 phase with a natural tubular halloysite nanotube (HNT) with 10~30 nm inner diameter as support, ammonium thiomolybdate and nickel nitrate were selected as metal precursors, and incipient-wetness impregnation method was applied to prepare a series of bimetallic NiMo/HNT catalysts. The structure of the as-prepared catalysts has been characterized with XRD, BET, HRTEM-EDS mapping and XPS. The desulfurization activity of the catalyst was evaluated with dibenzothiophene ( DBT ) as a model compound. It is found that the dimensions of the active MoS2 phase can be effectively controlled with the HNT support. Almost all the Mo and Ni metal components in the catalysts are located inside the pores of the support and present a native intimate contact state. Meanwhile, Ni and Mo components show a higher sulfidation degree and the synergy effect is better. The hydrodesulfurization activity and hydrogenation selectivity of the NiMo/HNT catalysts are significantly higher than those of the reference catalyst using typical alumina support. The highest DBT conversion, reaction rate and hydrogenation selectivity can be be increased up to 41.6 %, 92 % and 60%, respectively.
Keywords: hydrodesulfurization; active phase; halloysite nanotubes; spatial confinement strategy
馏分油加氢脱硫(HDS)催化剂的活性组分多为二元乃至多元金属复配组成,其中以VIB族Mo或W的硫化物为主剂,以VIII族的Co或Ni的硫化物为助剂[1-2]。负载型MoS2基HDS催化剂的活性主要取决于其MoS2加氢活性相的结构设计[3]。研究[4]表明,MoS2活性相的片层长度应在2~6 nm间集中分布且具有适当堆垛层数,助剂Ni中心需位于在MoS2活性相的边缘或邻近位置以保障双金属中心的协同效应。通过制备方式有效调控所得MoS2活性相的结构尺寸,同时保证Ni组分与MoS2中心之间的亲密接触,可定向制备高活性的负载型NiMo双组份HDS催化剂,因此具有特定孔道结构载体(如分子筛、碳纳米管等)的孔道限域效应引人注目[5]。其中具有纳米级管腔结构的天然黏土矿物埃洛石纳米管(HNT)极具优势[6-7]。目前已有CoMo/HNT-Al2O3催化剂应用于二苯并噻吩(DBT)HDS反应的报道[8],其HDS反应的TOF(磷酸三辛酯 )值优于使用Al2O3载体的参比剂,但是DBT转化率、加氢选择性等主要性能却劣于Al2O3载体的参比剂。基于柴油馏分HDS催化剂对脱硫活性和加氢选择性的更高要求,笔者使用具有更高加氢活性的NiMo基双金属催化剂体系,同时采用硫化态的Mo前驱物种硫代钼酸铵(ATM)进一步提升催化剂的加氢选择性[9]。
1 试 验
1.1 催化剂制备
催化剂的加氢活性组分选择NiMo双金属,分别以六水合硝酸镍和硫代钼酸铵作为催化剂的镍源和钼源,采用等体积浸渍法负载金属组分。浸渍前取适量事先经过空气气氛550 ℃焙烧4 h的HNT载体测定其吸水率,以此数据计算浸渍液中所含不同金属组分的质量。
(1)催化剂前体。以一定量的硫代钼酸铵ATM与硝酸镍溶解于水中配成所需浓度的浸渍液,使用等体积浸渍法浸渍到经过焙烧处理的HNT载体上,而后于60 ℃下真空干燥12 h,得到催化剂前体。使用传统Al2O3载体的参比催化剂,其制备工艺与HNT载体催化剂的相同,只是更换了氧化物载体。本文中使用的Al2O3载体以大孔氢氧化铝干胶为前驱体,经空气气氛550 ℃焙烧4 h得到。
(2)催化剂硫化。取一定量催化剂前体置于石英舟中于管式炉中硫化,硫化气氛为10% H2S/H2(体积比),硫化条件为400 ℃下恒温4 h。降温后取出硫化态催化剂,将其置于乙醇中保存备用或者用于其他表征以及HDS活性评价。
采用的催化剂命名规则为wxNiMS/support,其中MS表示以硫化态化合物(四硫代钼酸铵)为前驱体,w表示金属负载量(w=5%、10%、15%、20%),x表示Ni/Mo原子比(x=0.25、0.5、0.75、1),support为采用的载体(HNT载体或参比剂的Al2O3载体)。
1.2 催化剂表征
采用荷兰帕纳科公司生产的Empyrean锐影型X射线衍射仪(XRD)进行样品的物相分析,测定条件为衍射源Cu-Ka(λ=0.154 06 nm),管压40 kV,管流40 mA,接收狭缝0.3 mm,发散狭缝1 °,扫描速度15 °/min,扫描范围2θ为5 °~75 °。
采用美国麦克公司的Tristar-3020型物理吸附仪进行测定,测试前样品于150 ℃条件下真空脱气处理12 h。在77 K进行低温氮气吸脱附测试,用BJH方法计算得到催化剂的孔径和孔容数据,利用BET方法计算出催化剂比表面积。
采用美国赛默飞世尔公司的ESCALab 250Xi型X射线光电子能谱仪(XPS)采集催化剂样品的XPS数据。激发光源为AlKα(1 486.6 eV)射线,同时依照污染碳峰的C 1s(284.6 eV)的标准进行调试校准。Mo 3d、Ni 2p的XPS光谱使用XPSPeak软件进行拟合,采用Shirley背景扣除法,谱线分峰拟合时峰型使用高斯(80%)-洛伦兹(20%)的参数。
采用美国赛默飞世尔公司生产的Talos-F200X型场发射扫描透射电子显微镜(TEM)观察催化剂MoS2活性相的分布及微观形貌,采用仪器自带的EDS能谱仪进行催化剂微区元素组成的mapping分析,测试时电镜加速电压200 kV。TEM制样方式为将样品在乙醇中充分研磨后滴于表面覆盖碳膜的铜网上。本文中对MoS2活性相以GMS软件进行定量统计分析,样本数不低于150个计算得到不同催化剂中MoS2活性相的平均堆垛层数
()与平均片层长度()。
MoS2微晶的平均长度计算式为
=∑ti=1…tLit .(1)
式中,t为TEM统计得到的总样品数。
平均堆积层数可通过下式计算:
=∑ti=1…txiNi∑ti=1…txi .(2)
式中,xi为堆积层数为Ni的微晶数量。
1.3 催化剂活性评价
催化剂加氢脱硫评价在100 mL Easychem微型高压反应釜中进行,将0.5 g硫化态催化剂和40 mL质量分数为2%的DBT/十二烷溶液先后加入到100 mL高压釜中,使用可燃性气体检测仪确定装置气密性良好后,用H2连续置换5次以排除高压釜内的残余空气,初压为2 MPa,高压釜内反应温度设为320 ℃(反应温度下系统总压力为4 MPa),反应器搅拌速度为700 r/min,反应4 h后取样分析。使用Bruker456-GC型气相色谱仪对反应后的液相产物进行组成分析。
DBT加氢脱硫反应产物主要为4H-DBT、6H-DBT、BP和CHB。
(1)HDS转化率:
CDBT=1-c(DBT)c(DBT)+c(4H-DBT)+c(6H-DBT)+c(CHB)+c(BP) .(3)
(2)反应速率:
RDBT=nwCDBTt .(4)
(3)反应路径选择性:
SHYD/DDS=c(4H-DBT)+c(6H-DBT)+c(CHB)c(BP).(5)
式中,CDBT为DBT的转化率,%;c(DBT)、c(4H-DBT)、c(6H-DBT)、c(CHB)、c(BP)分别为反应产物中的二苯并噻吩(DBT)、四氢二苯并噻吩(4H-DBT)、六氢二苯并噻吩(6H-DBT)、环己基苯(CHB)、联苯(BP)的质量浓度;RDBT为催化剂的反应速率,mol·g-1·s-1;n为反应原料DBT的初始物质的量,mol;w为催化剂中活性组分MoS2的质量;t为反应时间;SHYD/DDS为反应路径选择性。
2 结果分析
采用特定制备方法制备系列NiMo/HNT催化剂,并采用多种分析手段对其结构性质进行表征,确认HNT载体孔道限域效应的存在。通过以二苯并噻吩(DBT)为模型化合物的HDS反应评价,系统考察不同制备工艺所得NiMo/HNT催化剂的HDS活性差异。
2.1 催化剂结构表征
2.1.1 X射线衍射
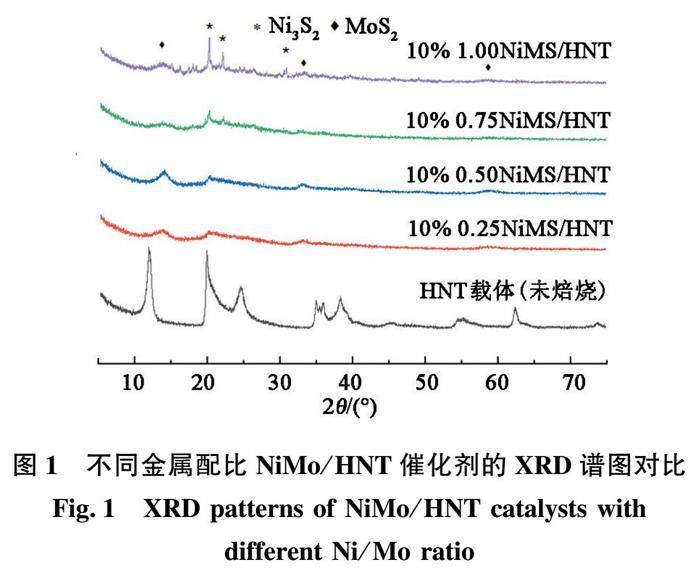
图1和图2分别为不同Ni/Mo原子比、不同金属总负载量的硫化态NiMS/HNT催化剂的XRD谱图。由图1可见,未经焙烧的HNT载体(分子式为Al2O3·2SiO2·4H2O)具有较好的结晶性能,而焙烧之后各催化剂中的HNT载体均呈现无定型Si-Al复合氧化物的状态;而金属组分方面除了有较强的归属于MoS2纳米颗粒的XRD特征衍射峰之外,当Ni/Mo较高(大于0.75)时还检测到了较强的归属为Ni3S2晶相的XRD衍射峰(JCPDS标准卡片编号[01-076-1870][10])。类似的结果在图2中也可见到,当催化剂中金属总负载量超过15%后,MoS2组分的结晶性能、分散程度基本没有变化,而Ni组分的衍射峰则逐渐明显。
XRD结果表明,当NiMo/HNT催化剂中Ni组分含量较高时,其分散均匀性下降,会出现明显聚集现象,而对于负载型NiMo基HDS催化剂而言,助剂Ni的存在以Ni位于MoS2片层边缘或附近而形成所谓的“Ni-Mo-S相”时催化剂的活性最好,此时Ni中心呈高分散状态,而不应该与MoS2形成分相[12]。推测该现象的原因,很可能是由于本文中采用的催化剂浸渍工艺是等体积一步浸渍法,当浸渍液中Ni组分含量(相对含量或者总含量)较高时,由于Ni、Mo前驱物种自身溶解度所导致的浸渍液的浓度及黏度较大,以及HNT载体自身特性(如纳米管长径比)等因素影响,不利于Ni中心在HNT载体上的分散,进而影响所得催化剂的最终活性。后面的反应结果也证实,当Ni、Mo两相形成分相时,催化剂的HDS活性有一定程度的下降。
2.1.2 低温N2吸脱附
表1和表2中分别对比了不同Ni/Mo原子比、不同金属总负载量的硫化态NiMo/HNT催化剂的孔结构分析数据,图3、4分别是对应的吸脱附等温线和孔径分布曲线。
由表1、表2中催化剂孔道结构数据可以发现,HNT载体引入Ni、Mo双金属组分之后,随着Ni/Mo(原子比)增大以及金属总负载量的增加,所得NiMo/HNT催化剂的比表面、孔体积、平均孔径等均呈明显的下降趋势,这表明金属组分已进入HNT载体的孔道之中。
图3、4分别为不同Ni/Mo原子比和不同金属总负载量的NiMS/HNT催化剂的吸脱附等温线和孔径分布曲线。对比可以发现,不论采用何种Ni/Mo比以及金属负载量,所得NiMo/HNT催化剂均有H3型滞后环与III型吸脱附等温线,证实了其介孔结构依然保持,而孔道结构更多的为大孔且相对更不规则;同时孔径分布曲线显示了催化剂孔道除了在10 nm及更大孔径区间有较多分布之外,在2 nm附近还有少量小介孔出现。
2.1.3 X射线光电子能谱(XPS)
采用X射线光电子能谱(XPS)表征手段对硫化态的NiMo/HNT及NiMo/Al2O3参比催化剂中Ni、Mo组分的价态进行了表征。以污染碳峰C 1s(284.8 eV)进行谱峰校准,采用XPSPEAK41软件分别对Ni 2p和Mo 3d谱峰进行分峰拟合,结果见图5。
Mo元素的拟合遵循以下规则[13-14]:每个价态的Mo 3d轨道分裂成Mo 3d5/2和Mo 3d3/2一对峰,两峰间距一般为3.1~3.2 eV,两峰的半峰宽(FWHM)相等,同价态的Mo 3d5/2和Mo 3d3/2两峰面积之比为3∶2,S 2s(BE=226±0.2 eV)对应的峰面积需被扣除。Mo 3d的第一组双峰出现在228.8 eV(Mo 3d5/2)及231.9 eV(Mo 3d3/2),归属于Mo(IV),即完全硫化后的Mo;第二组双峰出现在229.8 eV(Mo 3d5/2)和232.9 eV(Mo 3d3/2),归属于Mo(V),即未被完全硫化的氧硫化物中间态;第三组双峰出现在233.0 eV(Mo 3d5/2)和236.1 eV(Mo 3d3/2),归属于Mo(VI),即未被还原硫化的部分。三个峰位置均有峰表明催化剂存在不同程度硫化且不同价态的Mo。由图5对比可见,使用HNT载体有利于更高比例Mo(IV)组分形成(比例由使用Al2O3载体参比剂的50 %提升至90 %),即有利于更多MoS2活性相的形成,从而必将有利于催化剂HDS活性的提升。
在Ni物种价态解析的过程中,最关键区域的Ni 2p峰被分解为三个,分别对应Ni-S相(Ni2S3、Ni9S8或NiS,结合能约为852 eV)、与氧化铝相互作用的氧化态Ni2+(约为856 eV)以及NiMoS相(约为854 eV)[15],同时为了完成对Ni元素的精确分析,根据三个主峰的结合能和区域关系,还添加了Ni的三个卫星峰。XPS结果表明,虽然使用HNT载体时催化剂中的Ni物种也无法完全硫化,但其价态分布中NiMoS活性相的比例相对Al2O3负载的参比剂有明显升高(由27.8 %提升至74.3 %),这表明使用HNT载体可以提高NiMo催化剂中Ni、Mo组分之间的协同作用,其优势也将从HDS活性评价数据得到印证。
2.1.4 高分辨透射电子显微镜(HRTEM)
反应后的硫化态负载型NiMo催化剂的HRTEM及EDS mapping表征结果如图6所示。相关形貌参数的统计结果见表3所示。
由图6(a)、(b)中NiMS/HNT催化剂的HRTEM图像可见,NiMo双金属催化剂中的MoS2活性相的堆垛清晰,且基本上均位于孔道内部,HNT载体外表面几乎没有MoS2物相存在。由图6(c)~(i)中EDS mapping结果能够看出,10% 0.5NiMS/HNT催化剂中的Ni、Mo、S元素分散均匀,且基本全部位于孔道之中,这表明使用HNT载体的孔道限域效果十分明显。
对比表3中NiMo/HNT以及NiMo/Al2O3参比催化剂中MoS2活性相的片层长度和堆垛层数的形貌参数可见,使用HNT载体可显著提高催化剂中MoS2活性相的堆垛层数,同时片层长度也有一定程度的增加,这很可能是由于焙烧后的HNT载体为无定型Si-Al复合氧化物,其与Mo组分作用力更弱,同样条件下Mo组分的硫化程度更高,因而片晶结构尺寸整体有所增大,这也与XPS的表征数据相互印证。其中MoS2活性相堆垛数提升将对催化剂的加氢活性具有积极意义,根据Topse等[11]提出的“Ni-Mo-S加氢活性相”模型,即在加氢催化剂中存在Type I型和Type II型活性相,其中II型活性相因与载体相互作用弱导致硫化度高、堆积层数高,相比I型活性相其脱硫性能较高。因此MoS2活性相堆垛层数的提高将与催化剂加氢活性及整体HDS活性的提高成正相关,这一点也可从后面的HDS反应性能评价结果得到证实。
由此可见,HNT载体的意义除了孔道限域效应在空间上对MoS2活性相的结构尺寸、Ni-Mo双组分的分布存在位置等方面产生了控制影响之外,还因为其载体-MoS2组分之间的作用力相对更弱,更有利于高活性II型活性相的形成,将对所得催化剂整体HDS活性的提升具有积极意义。
2.2 催化剂的HDS活性评价
为验证HNT载体的使用效果,以二苯并噻吩(DBT)为模型化合物,考察不同制备工艺(不同Ni/Mo比,不同金属负载量)的NiMo/HNT催化剂以及使用Al2O3载体的参比剂的HDS活性,相关结果见表4。
由表4中HDS反应评价结果可见以下规律:①相比使用Al2O3载体的参比剂,NiMo/HNT催化剂的HDS活性有大幅增加(反应速率R最大可以提升92 %,DBT转化率最高可提升41.6 %),同时加氢选择性也有明显提升(最大可提升60 %);②对比不同Ni/Mo原子比的NiMo/HNT催化剂,以Ni/Mo为0.5的活性最好(转化率、反应速率、加氢活性),Ni/Mo比超过0.5后催化剂的加氢活性和HDS活性反而下降;③对比不同金属负载量的NiMo/HNT催化剂,以负载量10%~15%的催化剂活性转化率最好,但如果以基于单位质量MoS2用量的转化率反应速率R考虑,5%的用量更优,催化剂金属负载量进一步提高不利于催化剂整体活性的提升,这一点与图1和图2中XRD表征反应的高负载量时Ni、Mo组分发生分相的现象相吻合。其本质原因可以根据密度泛函理论(DFT)计算出的硫化态活性组分的金属-硫键键能(EMS)来解释[16-17]:相比分相形态的Ni3S2和MoS2的EMS,更优的存在形态为“Ni-Mo-S”相,其EMS值大小适中更有利于加氢脱硫反应的发生。所以如果制备条件不合适,将导致催化剂中的Ni组分与MoS2组分处于分相形态时,所得NiMo催化剂的整体活性将受到较大影响,需在催化剂制备工艺中注意避免。
3 结 论
(1)以具有内径为10~30 nm直通孔道的天然埃洛石纳米管(HNT)为载体,以硫代钼酸铵和硝酸镍为金属前驱物,采用合适的工艺条件制备的系列双金属NiMo/HNT催化剂,可以实现载体结构对MoS2加氢活性相的“孔道空间限域效应”。
(2)HNT负载催化剂中MoS2活性相的平均长度控制在小于6 nm,平均堆垛层数相比传统使用Al2O3载体的参比催化剂有明显提升,同时催化剂中的Ni、Mo组分的硫化程度更高且分布均匀,有效保障了双金属中心的协同效应。
(3)采用优化后的制备工艺,所得的NiMo/HNT催化剂的加氢脱硫活性和加氢选择性均显著优于使用常规Al2O3载体的参比催化剂,其中反应速率R最大可以提升92%,DBT转化率最高可提升41.6%,加氢选择性最大可提升60 %。
参考文献:
[1] 孙昱东,魏成,韩忠祥,等.临氢缓和条件下煤-油共炼过程反应机制[J].中国石油大学学报(自然科学版),2022,46(3):174-179.
SUN Yudong, WEI Cheng, HAN Zhongxiang, et al. Mechanism of coal oil co-refining under mild condition in hydrogen[J]. Journal of China University of Petroleum(Edition of Natural Science),2022,46(3):174-179.
[2] EGOROVA M, PRINS R. Hydrodesulfurization of dibenzothiophene and 4,6-dimethyldibenzothiophene over sulfided NiMo/γ-Al2O3, CoMo/γ-Al2O3, and Mo/γ-Al2O3 catalysts[J]. Journal of Catalysis, 2004, 225(2):417-427.
[3] DELMON B, FROMENT G F. Remote control of catalytic sites by spillover species: a chemical reaction engineering approach[J]. Catalysis Reviews: Science and Engineering, 1996, 38(1):69-100.
[4] LAURITSEN J V, BOLLINGER M V, LAEGSGAARD E, et al. Atomic-scale insight into structure and morphology changes of MoS2 nanoclusters in hydrotreating catalysts[J]. Journal of Catalysis, 2004, 221(2):510-522.
[5] OKAMOTO Y, KUBOTA T. A model catalyst approach to the effects of the support on Co-Mo hydrodesulfurization catalysts[J]. Catalysis Today, 2003, 86(1):31-43.
[6] LVOV Y, WANG W, ZHANG L, et al. Halloysiteclay nanotubes for loading and sustained release of functional compounds[J]. Advanced Materials, 2016, 28(6):1227-1250.
[7] MASSARO M, COLLETTI C G, LAZZARA G, et al. Halloysite nanotubes as support for metal-based catalysts[J]. Journal of Materials Chemistry A, 2017, 5(26):13276-13293.
[8] PIMERZIN A A, VUTOLKINA A V, VINOGRADOV N A, et al. Core-shell catalysts with CoMoS phase embedded in clay nanotubes for dibenzothiophene hydrodesulfurization[J]. Catalysis Today, 2022,397/398/399:121-128.
[9] HUANG Z D, BENSCH W, KIENLE L, et al. SBA-15 assupport for Ni-MoS2 HDS catalysts derived from sulfur-containing molybdenum and nickel complexes in the reaction of HDS of DBT: an all sulfide route[J]. Catalysis Letters, 2009,127(1):132-142.
[10] PALCHEVA R, KALUA L, DIMITROV L, et al. NiMo catalysts supported on the Nb modified mesoporous SBA-15 and HMS:effect of thioglycolic acid addition on HDS[J]. Applied Catalysis A: General, 2016,520:24-34.
[11] TOPSOE H, CLAUSEN B S, CANDIA R, et al. In situ Mssbauer emission spectroscopy studies of unsupported and supported sulfided Co-Mo hydrodesulfurization catalysts:evidence for and nature of a Co-Mo-S phase[J]. Journal of Catalysis, 1981,68(2):433-452.
[12] SANTOLALLA-VARGAS C E, SUáREZ TORIELLO V A, DE LOS REYES J A, et al. Effects of pH and chelating agent on the NiWS phase formation in NiW/γ-Al2O3 HDS catalysts[J]. Materials Chemistry and Physics, 2015,166:105-115.
[13] WU H B, XIA B Y, YU L, et al. Porous molybdenum carbide nano-octahedrons synthesized via confined carburization in metal-organic frameworks for efficient hydrogen production[J]. Nature Communications, 2015,6(1):6512.
[14] TANG Y J, GAO M R, LIU C H, et al. Porous molybdenum-based hybrid catalysts for highly efficient hydrogen evolution[J]. Angewandte Chemie International Edition, 2015,54(44):12928-12932.
[15] NESBITT H W, LEGRAND D, BANCROFT G M. Interpretation of Ni 2p XPS spectra of Ni conductors and Ni insulators[J]. Physics and Chemistry of Minerals, 2000,27(5):357-366.
[16] LAURITSEN J, KIBSGAARD J, OLESEN G, et al. Location and coordination of promoter atoms in Co- and Ni-promoted MoS2-based hydrotreating catalysts[J]. Journal of Catalysis, 2007,249(2):220-233.
[17] DINTER N, RUSANEN M, RAYBAUD P, et al. Temperature-programed reduction of unpromoted MoS2-based hydrodesulfurization catalysts: experiments and kinetic modeling from first principles[J]. Journal of Catalysis, 2009,267(1):67-77.
(编辑 刘为清)
基金项目:中央高校基本科研业务费专项(22CX01002A)
第一作者及通信作者:李彦鹏(1979-),男,副教授,博士,硕士生导师,研究方向为加氢催化剂及电子显微学。E-mail: liyanpeng@upc.edu.cn。
引用格式:李彦鹏,孙于林,杨兆胜,等.负载型MoS2基加氢催化剂活性相的限域构建及其加氢脱硫活性[J].中国石油大学学报(自然科学版),2024,48(3):207-214.
LI Yanpeng, SUN Yulin, YANG Zhaosheng, et al. Active-phase construction and hydrodesulfurization activity evaluation of supported MoS2-based hydrogenation catalysts designed with spatial confinement strategy[J].Journal of China University of Petroleum(Edition of Natural Science),2024,48(3):207-214.
