DUT-4选择性吸附SO2/CO2混合气体中SO2的理论研究
2024-06-16黄啸翔倪超
黄啸翔 倪超

摘 要:金属有机骨架(MOFs)作为吸附剂选择性捕获SO2是一种有前景的烟气脱硫技术。采用密度泛函理论(DFT)计算和巨正则蒙特卡罗(GCMC)模拟,研究铝基MOFs材料DUT-4对纯SO2和CO2的吸附机理,并探究其对SO2/CO2混合气体的吸附选择性。结果表明:由于金属中心的高静电势梯度和羟基中Hμ-oh与气体分子间的氢键作用,使得DUT-4吸附剂中SO2与CO2主要吸附在靠近金属中心的位置。低温、高压、高SO2比例均会提高DUT-4对烟气中SO2的吸附选择性。
关 键 词:DFT; GCMC; 金属有机框架; SO2/CO2; 选择性吸附
中图分类号:TQ424 文献标识码: A 文章编号: 1004-0935(2024)05-0691-06
化石燃料燃烧过程中排放的CO2与SO2等酸性气体对环境与人体健康有极大危害。目前,最为成熟的CO2排放控制技术是化学吸附法,通过有机胺和CO2发生化学反应从而回收并分离CO2[1]。然而烟气中的SO2会使有机胺失去活性,不利于CO2的捕集[2]。因此开发高效、经济的SO2捕集及分离技术十分必要[3]。
目前,烟气脱硫主要分为湿法脱硫和干法脱硫。湿法脱硫有石灰/石灰石烟气脱硫法、氨法烟气脱硫法等,其脱硫效率达90%以上,具有脱硫效率高、投资费用低等优势[4],但同时也存在处理过程产生大量废水、设备维护繁琐等缺点[5]。干法脱硫有循环流化床脱硫法等方法,工艺简单,设备维护方便,但脱硫效率较低[6]。尽管这些脱硫方法是有效的,但燃煤烟气中仍有残留SO2,排放到大气中时,依然会造成健康风险和环境危害。
金属有机框架(MOFs)是一种由金属中心和有机连接体自组装而成的三维网状多孔材料[7-8]。由于其巨大的表面积、可调节孔径和可调控的表面性能,MOFs在分离、催化和气体储存方面具有潜在的应用前景[9-11]。现阶段很多工作都集中在研究MOFs中的气体分离[12-16],但很少有人开展将其用于SO2封存的研究,因为它经常导致材料的严重结构退化或不可逆的吸收。DUT-4是一种中心金属为Al(Ⅲ)、通过2,6-萘二甲酸 (2,6-NDA)连接的MOFs,具有良好的SO2吸附性能、良好的循环性、高热稳定性及易吸附可逆性,是作为烟气脱硫非常有前途的气体存储和分离材料[17]。
本文通过密度泛函理论(DFT)计算和巨正则蒙特卡罗(GCMC)模拟方法研究SO2、CO2以及SO2/CO2混合气体在DUT-4孔道内的吸附机理,并对混合气体的吸附选择性进行了预测,探讨了温度、压力及混合气体比例对SO2/CO2混合气体的选择性吸附影响。
1 计算方法
1.1 DFT计算细节
在VASP软件[18]中使用密度泛函理论(DFT)优化了DUT-4的晶胞结构,交换相关函数采用 PBE 的广义梯度近似(GGA)[19],计算中的截断能采用500 eV,K-Point设置为2×5×2,结构优化设置总能量变化小于10-5 eV,自洽计算时要求总能量变化小于10-6 eV,计算了DUT-4中的电荷密度与单个CO2分子和SO2分子在DUT-4中的吸附能。吸附能计算公式如式(1)所示。
(1)式中:Eads—气体在DUT-4上的吸附能;
Eg/DUT-4—吸附体系总能量;
Eg—气体能量;
EDUT-4—DUT-4的能量。
1.2 GCMC模拟细节
通过RASPA2.0软件[20],采用巨正则蒙特卡罗(GCMC)方法模拟了DUT-4在298 K下SO2、CO2的吸附和扩散特性,DUT-4框架采用Dreiding力 场[21]确定力场参数,CO2的分子模型的力场参数来源于PETERS[22]等的研究结果,SO2的分子模型取于KETKO[23]等的研究结果,模拟框架采用DUT-4的 2×4×2超胞,DUT-4超胞与气体小分子均视为刚性,原子间范德华力用12-6 LJ表达式描述,截断半径取1.3 nm。长距离静电相互作用力使用 Ewald求和来处理,具体参数如表1、表2所示。孔隙率设为0.66,这是计算得到的氦孔隙率,每个模拟总共执行106个生产循环步和106个平衡循环步。
气体在DUT-4中的吸附热(Qst)由公式(2)得出[24]。
(2)式中:U—总能量;
N—吸附分子的数量。
吸附选择性的计算公式如下[25]:
(3)式中:x、y—吸附质i、j在气相和吸附相中的摩尔分数。
2 结果与讨论
2.1 模型验证
为了验证DFT优化的DUT-4晶体模型(见 图1a)的可靠性,将VESTA[26]软件计算DUT-4结构模型得到的PXRD图与实验得到的PXRD图[17]进行比较(见图1b),模拟值与实验值的主峰位置和相对强度基本一致,证明了DUT-4结构模型的合理性,可以满足模拟要求。
通过GCMC对CO2、SO2气体的吸附等温线模拟对比实验结果以验证力场参数的可靠性,图1(c)为不同压力下CO2在273 K与303 K下的模拟与实验得到的吸附等温线[27]和不同压力下SO2在298 K时的模拟与实验得到的吸附等温线[17]。
(a)DUT-4结构模型
(b)DUT-4的PXRD图谱
(c)DUT-4的吸附等温线
(d)SO2与CO2吸附等温线
图1 DUT-4结构模型、报道中和本文模拟DUT-4的PXRD图谱、实验与模拟的气体在DUT-4的吸附等温线以及298 K下模拟的SO2与CO2吸附等温线
由图1中的曲线可以看出,本文模拟得到等温吸附曲线与实验数据基本相符,说明本文采用的参数可靠,可以用于研究DUT-4的选择性吸附行为。
2.2 单组分气体吸附
图1(d)显示了CO2与SO2在298 K的吸附等温线,低压下SO2的吸附量随压力增加较快,接近100 kPa时吸附增加量趋于平缓,而CO2的吸附量随压力增大呈线性增加,但增加缓慢,CO2在10 kPa与100 kPa下均远小于SO2的饱和吸附量。通过GCMC计算了2种气体在极低压力下的等量吸附热Qst,CO2的等量吸附热为-17.757 kJ·mol-1,SO2的等量吸附热为-24.707 kJ·mol-1,这表明DUT-4对于SO2有更高的吸附倾向。
吸附结构的快照被广泛用于研究MOFs的吸附性能,因此从分子水平计算了气体在DUT-4中的吸附结构的快照,模拟了温度298 K,压力0.01、0.1、100 kPa下,DUT-4分别对CO2与SO2的吸附快照,如图2所示。在1 kPa下,CO2与SO2在孔道中吸附剂较少。对于CO2,随着压力升高,达到100 kPa 时,各孔道出现CO2分子,但仍有大量空间。对于SO2,达到10 kPa时,SO2分子数量快速增加;压力增加到100 kPa 时,SO2几乎占满DUT-4的孔道。由图2可知,中高压下CO2吸附量增速几乎不变,而SO2吸附量增加缓慢,与图1(d)中的结果一致。
为确定CO2和SO2在DUT-4中吸附位置及作用机理,通过GCMC模拟了DUT-4中吸附质分子的分布,通过DFT计算了DUT-4框架结构的电荷密度、吸附质的吸附位点和吸附能,如图3所示,分别是CO2、SO2在1、100 kPa 下的吸附密度图,图中显示在1 kPa下CO2与SO2均在四边形孔径的4个接近金属中心的区域,而孔道中心区域的气体分子分布极少。随着压力增加,100 kPa时,SO2填满孔道角落,高密度分布区逐渐往孔道中心扩散。而CO2的吸附量尚未填满孔道角落,高密度分布区与低压时相似,CO2依然吸附在孔道中接近金属中心的位置。
(a)SO2(1 kPa ) (b)CO2(1 kPa )
(c)SO2(100 kPa ) (d)CO2(100 kPa )
(e)SO2在DUT-4上的径向分布函数
(f)CO2在DUT-4上的径向分布函数
(S_so2和C_co2分别表示为SO2和CO2中的硫和原子;Al、Cnap和Hμ-oh分别为DUT-4中的中心金属铝、连接体萘的中心碳原子和金属中心的羟基氢)
图3 298 K下1 kPa和100 kPa 时DUT-4吸附单组分SO2和CO2的分布密度图以及298 K和100 kPa下SO2和CO2在DUT-4上的径向分布函数
随后计算了CO2与SO2在DUT-4框架中的径向分布函数(RDF),如图3(e)和图3f所示,2种气体均在金属中心的羟基基团中氢原子距离0.2~0.3 nm附近处出现,表明羟基和2种气体间有弱的氢键相互作用,这在其他MOF吸附含氧气体小分子的研究中经常出现[28],距离连接体萘上的高密度吸附可能是由于CO2和SO2分子与芳香环的相互作用[29],距离中心金属Al较远的原因是Al原子被O原子包围,空间粒子密度较大,气体分子难以靠近。
图4(a)为DFT-4框架上的电荷密度图,红色区域代表电子密度较大,可以看出与有机配体相比,金属中心富集了大量电子,从而具有更大的静电势梯度,所以吸附质倾向于吸附在金属中心[30]。通过DFT计算了气体在DUT-4上的有利吸附位点与吸附能,在对应位点的SO2的吸附能为-0.577 eV,CO2的吸附能为-0.203 eV,SO2更负的吸附能也表明SO2比CO2更容易吸附在金属中心处,SO2与CO2中氧原子均与金属中心上羟基中的H原子形成氢键,氢键键长分别为0.219 0 nm和0.227 3 nm,这与径向分布函数的模拟结果相似,由于CO2与SO2相对于Al、Cnap和Hμ-oh分布距离相似,优势吸附区域重叠,2种气体间有吸附竞争关系,这就有利于SO2/CO2混合气体的选择性吸附。
(a)电荷密度图 (b)SO2有利吸附位点 (c)CO2有利吸附位点
2.3 双组分系统SO2的吸附选择性
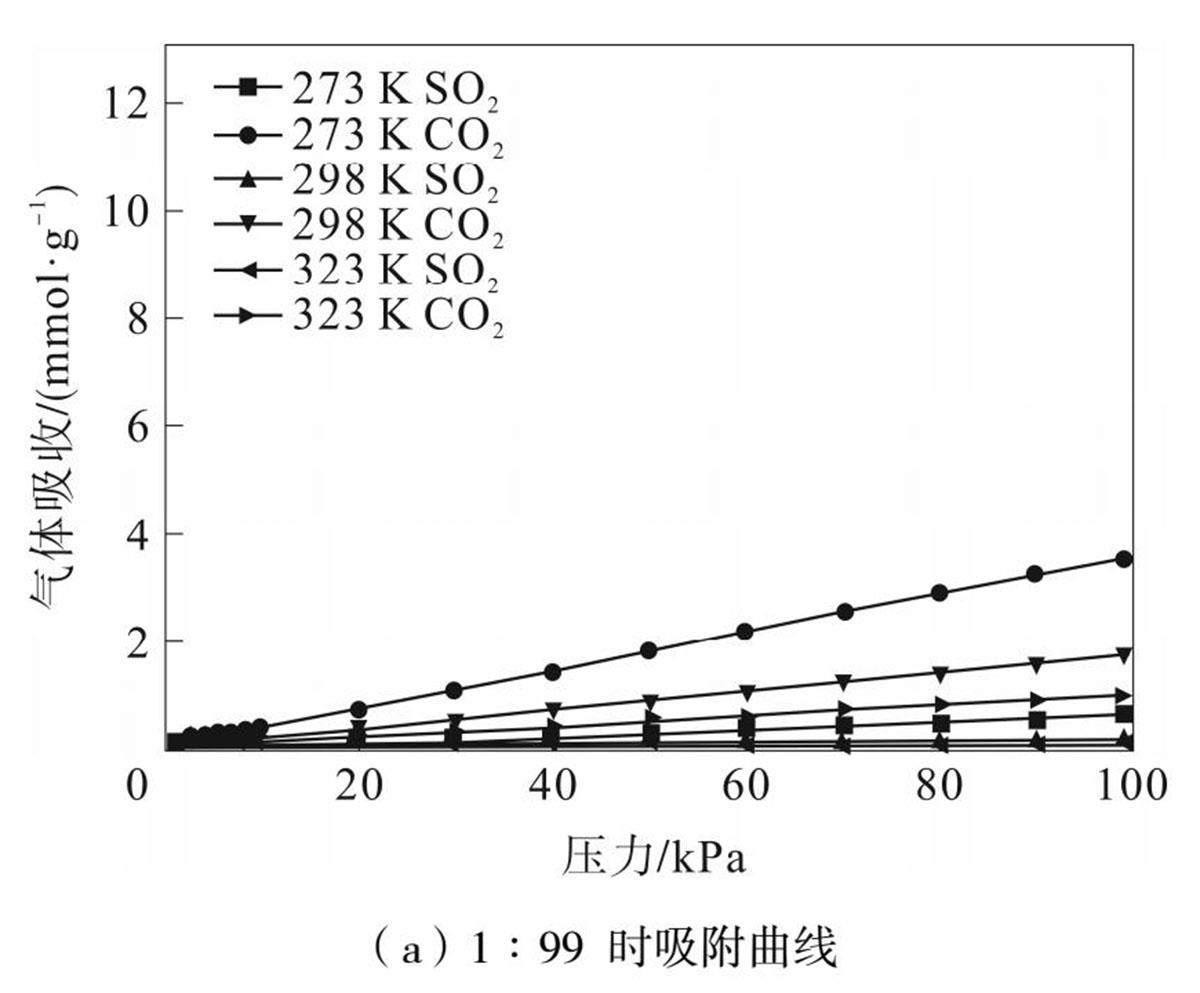
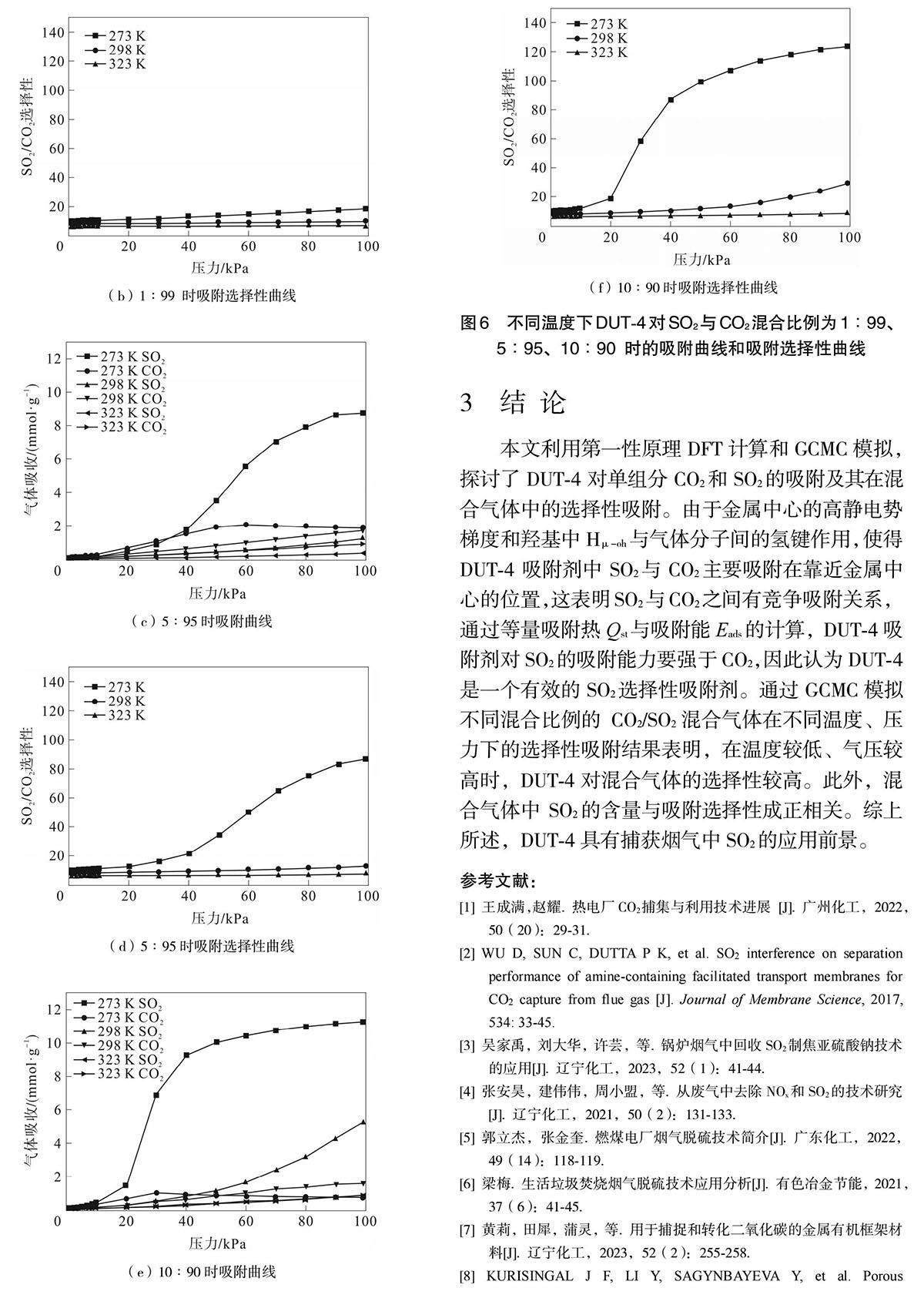
为了进一步研究DUT-4的选择性吸附性能,并探索其在工业过程中的潜在应用,通过GCMC预测了DUT-4对SO2/CO2混合气体的吸附选择性,模拟了DUT-4在不同温度(273、298、323 K)和不同比例(1∶99、5∶95、10∶90)的SO2/CO2混合气体的吸附等温线、吸附快照以及选择性吸附曲线,结果如图5、图6所示。
由图5和图6(a)、图6(c)、图6(e)可以得出,随着温度的升高混合气体的吸附量逐渐减小。在323 K时,SO2与CO2的吸附量随压强线性增加,这表明DUT-4孔道内的气体分子数量较少,还存在大量未被占据的吸附位点。温度降到273 K时,SO2吸附量随着压力上升逐渐饱和,同时SO2与CO2之间存在竞争吸附关系,在孔道趋于饱和时,由于DUT-4对SO2更强烈的吸附倾向,CO2的吸附量逐渐减小,这在图(c)、图6(e)中表现明显,这使DUT-4对混合气体中SO2的选择性提高。同理,混合气体的比例也影响了最终的气体分离效率,SO2与CO2比例为10∶90的混合气体的分离效率要高于5∶95和10∶90的混合气体。
图6(b)、 图6(d)、图6(f)展示了DUT-4在不同情况下的SO2/CO2混合气体吸附选择性,一般来说,高压下的SO2/CO2选择性要高,10∶90的SO2/CO2混合气体在273 K下,总压100 kPa 时的选择性为123.71,远大于总压10 kPa和1 kPa时的12.71和10.62,在较低压力下,孔道选择性变化不大。相同浓度与压力下,温度越高选择性越低,低压选择性变化较小时,323 K时选择性在6.5附近,而273 K下的SO2/CO2混合气体选择性在11附近。比例不同的CO2/SO2混合气体在较高压力时体现出选择性的差异。
(a)1∶99 时吸附曲线
(b)1∶99 时吸附选择性曲线
(c)5∶95时吸附曲线
(d)5∶95时吸附选择性曲线
(e)10∶90时吸附曲线
(f)10∶90时吸附选择性曲线
3 结 论
本文利用第一性原理DFT计算和GCMC模拟,探讨了DUT-4对单组分CO2和SO2的吸附及其在混合气体中的选择性吸附。由于金属中心的高静电势梯度和羟基中Hμ-oh与气体分子间的氢键作用,使得DUT-4吸附剂中SO2与CO2主要吸附在靠近金属中心的位置,这表明SO2与CO2之间有竞争吸附关系,通过等量吸附热Qst与吸附能Eads的计算,DUT-4吸附剂对SO2的吸附能力要强于CO2,因此认为DUT-4是一个有效的SO2选择性吸附剂。通过GCMC模拟不同混合比例的CO2/SO2混合气体在不同温度、压力下的选择性吸附结果表明,在温度较低、气压较高时,DUT-4对混合气体的选择性较高。此外,混合气体中SO2的含量与吸附选择性成正相关。综上所述,DUT-4具有捕获烟气中SO2的应用前景。
参考文献:
[1] 王成满,赵耀. 热电厂CO2捕集与利用技术进展 [J]. 广州化工, 2022,50(20):29-31.
[2] WU D, SUN C, DUTTA P K, et al. SO2 interference on separation performance of amine-containing facilitated transport membranes for CO2 capture from flue gas [J]. Journal of Membrane Science, 2017, 534: 33-45.
[3] 吴家禹,刘大华,许芸,等. 锅炉烟气中回收SO2制焦亚硫酸钠技术的应用[J]. 辽宁化工,2023,52(1):41-44.
[4] 张安昊,建伟伟,周小盟,等. 从废气中去除NOx和SO2的技术研究 [J]. 辽宁化工,2021,50(2):131-133.
[5] 郭立杰,张金奎. 燃煤电厂烟气脱硫技术简介[J]. 广东化工,2022,49(14):118-119.
[6] 梁梅. 生活垃圾焚烧烟气脱硫技术应用分析[J]. 有色冶金节能,2021, 37(6):41-45.
[7] 黄莉,田犀,蒲灵,等. 用于捕捉和转化二氧化碳的金属有机框架材料[J]. 辽宁化工,2023,52(2):255-258.
[8] KURISINGAL J F, LI Y, SAGYNBAYEVA Y, et al. Porous aluminum-based DUT metal-organic frameworks for the transformation of CO2 into cyclic carbonates: A computationally supported study [J]. Catalysis Today, 2020, 352: 227-236.
[9] MARTINEZ-AHUMADA E, DIAZ-RAMIREZ M L, VELASQUEZ- HERNANDEZ M J, et al. Capture of toxic gases in MOFs: SO2, H2S, NH3 and NOx [J]. Chem Sci, 2021, 12(20): 6772-6799.
[10] XIA L, BO Z, LIU Q, et al. Li-doped and functionalized metal-organic framework-519 for enhancing hydrogen storage: a computational study [J]. Computational Materials Science, 2019, 166: 179-186.
[11] SUN W, LI H, LI H, et al. Adsorption mechanisms of ibuprofen and naproxen to UiO-66 and UiO-66-NH2: Batch experiment and DFT calculation [J]. Chemical Engineering Journal, 2019, 360: 645-653.
[12] PHAM T, SPACE B. Insights into the gas adsorption mechanisms in metal-organic frameworks from classical molecular simulations [J]. Top Curr Chem (Cham), 2020, 378(1): 14.
[13] YU S, JING G, LI S, et al. Tuning the hydrogen storage properties of MOF-650: A combined DFT and GCMC simulations study [J]. International Journal of Hydrogen Energy, 2020, 45(11): 6757-6764.
[14] GU C, LIU Y, WANG W, et al. Effects of functional groups for CO2 capture using metal organic frameworks [J]. Frontiers of Chemical Science and Engineering, 2020, 15(2): 437-449.
[15] MA X, LIU B, WU Q, et al. Specific Li+ sites in a nanoporous carbon for enhanced light hydrocarbons storage and separation: GCMC and DFT simulations [J]. Fuel, 2021, 288: 119647.
[16] SOKHANVARAN V, GOMAR M, YEGANEGI S. H2S separation from biogas by adsorption on functionalized MIL-47-X (X = ?OH and ? OCH3): a simulation study [J]. Applied Surface Science, 2019, 479: 1006-1013.
[17] L?PEZ-OLVERA A, PIOQUINTO-GARCíA S, ANTONIO ZáRATE J, et al. SO2 capture in a chemical stable Al(III) MOF: DUT-4 as an effective adsorbent to clean CH4 [J]. Fuel, 2022, 322: 124213.
[18] KRESSE G, FURTHMüLLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set [J]. Computational Materials Science, 1996, 6(1): 15-50.
[19] SEGALL M D, PHILIP J D L, PROBERT M J, et al. First-principles simulation: ideas, illustrations and the CASTEP code [J]. Journal of Physics: Condensed Matter, 2002, 14(11): 2717.
[20] DUBBELDAM D, CALERO S, ELLIS D E, et al. RASPA: molecular simulation software for adsorption and diffusion in flexible nanoporous materials [J]. Molecular Simulation, 2015, 42(2): 81-101.
[21] MAYO S L, OLAFSON B D, GODDARD W A. DREIDING: a generic force field for molecular simulations [J]. The Journal of Physical Chemistry, 1990, 94(26): 8897-909.
[22] PETERS S, RENJITH PILLAI S, VARATHAN E. Molecular simulations to investigate the guest-induced flexibility of Pu-UiO-66 MOF [J]. Materials Today: Proceedings, 2022, 68: 35-42.
[23] KETKO M H, KAMATH G, POTOFF J J. Development of an optimized intermolecular potential for sulfur dioxide [J]. J Phys Chem B, 2011, 115(17): 4949-4954.
[24] BABARAO R, HU Z, JIANG J, et al. Storage and Separation of CO2 and CH4 in Silicalite, C168 Schwarzite, and IRMOF-1: A Comparative Study from Monte Carlo Simulation [J]. Langmuir, 2007, 23(2): 659-666.
[25] CAO D, WU J. Modeling the selectivity of activated carbons for efficient separation of hydrogen and carbon dioxide [J]. Carbon, 2005, 43(7): 1364-1370.
[26] MOMMA K, IZUMI F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data [J]. Journal of Applied Crystallography, 2011, 44(6): 1272-1276.
[27] WANG G B, LEUS K, HENDRICKX K, et al. A series of sulfonic acid functionalized mixed-linker DUT-4 analogues: synthesis, gas sorption properties and catalytic performance [J]. Dalton Trans, 2017, 46(41): 14356-14364.
[28] LIU J X, LI J, TAO W Q, et al. 3.Al-based metal-organic framework MFM-300 and MIL-160 for SO2 capture: a molecular simulation study [J]. Fluid Phase Equilibria, 2021, 536: 112963.
[29] TORRISI A, MELLOT‐DRAZNIEKS C, BELL R G. Impact of ligands on CO2 adsorption in metal-organic frameworks: First principles study of the interaction of CO2 with functionalized benzenes. II. Effect of polar and acidic substituents [J]. The Journal of chemical physics, 2010, 132 4: 044705.
[30] XU Q, LIU D, YANG Q, et al. Li-modified metal–organic frameworks for CO2/CH4 separation: a route to achieving high adsorption selectivity [J]. J Mater Chem, 2010, 20(4): 706-714.
Theoretical Investigation on SO2/CO2 Selective Adsorption of DUT-4
HUANG Xiaoxiang1,2, NI Chao1,2
(1. Key Laboratory of Coal Processing and Efficient Utilization, Ministry of Education, Xuzhou Jiangsu 221116, China;
2. School of Chemical Engineering and Technology, China University of Mining and Technology, Xuzhou Jiangsu 221116, China)
Abstract: The emission of SO2from fossil fuel combustion without treatment will lead to various environmental and health hazards. The selective capture of SO2using (Metal-organic framework) MOFs as adsorbent is a promising technology for flue gas desulfurization. In this paper, first-principles density functional theory (DFT) calculation and Grand Canonical Monte Carlo (GCMC) simulation were used to study the adsorption mechanism of Al based MOFs material DUT-4 on pure SO2 and CO2, and the adsorption selectivity of SO2/CO2mixture gas was explored. It was found that low temperature, high pressure, high SO2ratio could improve the selectivity of SO2in flue gas.
Key words: DFT; GCMC; Metal-organic frame; SO2/CO2; Selective adsorption
收稿日期: 2023-03-20
作者简介: 黄啸翔(1998-),男,江苏省盐城市人,在读研究生,研究方向:理论计算。
通信作者: 倪 超(1988-),男,讲师,博士,研究方向:煤炭高效清洁利用,煤炭浮选。
