一氧化氮与哺乳动物卵母细胞成熟
2024-06-03李亚杰雷安民
李 潇,李亚杰,刘 珊,雷安民
(西北农林科技大学动物医学院,陕西杨凌 712100)
卵母细胞发生发育是多种细胞因子调节的结果,一氧化氮(nitric oxide,NO)作为一种具有自由基性质的多功能活性分子,参与了卵泡发生、卵母细胞减数分裂成熟、卵泡排卵、卵泡黄体化以及卵泡闭锁等重要过程。研究证明,一氧化氮合酶(nitric oxide synthase,NOS)在多物种生殖系统中表达,NO经过cGMP/PDE/Cdc25B/MPF通路触发多数哺乳动物卵母细胞的生发泡破裂(germinal vesicle breakdown,GVBD)及第二次减数分裂中期(MⅡ)停滞。因此,合理利用NO对卵母细胞减数分裂发育的激发性、抑制性提高卵母细胞质量,是一项值得深入研究的课题。本文就NO与卵母细胞成熟进程的关系进行介绍,以期为研究临床提高卵母细胞质量的NO相关培养方法及药物提供参考。
1 NO的发现及相关信号通路
20世纪70年代后期,Murad等研究发现硝酸甘油这一强力血管舒张剂通过释放NO气体分子引起血管平滑肌细胞内组织环磷酸鸟苷(cGMP)水平升高,促使心血管舒张,实现降低血压和疏通心脏冠状血管的生物作用。1980年,Furchgott发现血管内皮细胞释放一种可引起血管平滑肌舒张的弥散性物质,并将其命名为内皮源性舒张因子(endothelium-derived relaxing factor,EDRF);直到1986年,EDRF被确认为一氧化氮(NO),科学界正式开始了NO在心血管系统方面的功能研究。Furchgott等也因为关于NO在血管舒张中的开创性研究工作分享了1998年诺贝尔生物与医学奖。现在,硝酸甘油作为血管扩张类药物被广泛应用于现代医学领域,近年来,人们对NO在组织细胞中的研究也扩展到了免疫、代谢、神经和生殖等研究领域。
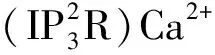
2 动物机体内NO的来源及产生通路
NO通过生物体内多种细胞类型的酶和非酶途径产生。酶途径的NO由一氧化氮合酶(NOS)催化产生,由于胞外NO的半衰期非常短而且NOS是一氧化氮生成过程中的限速酶,同时NO没有专门的贮存及调节释放机制,因此某一组织中NOS的量能线性反应出NO的浓度水平。哺乳动物中NOS包括神经元型一氧化氮合酶(neuronal nitricoxidesynthase,nNOS)、内皮型一氧化氮合酶(endothelial nitricoxidesynthase,eNOS)和诱导性一氧化氮合酶(inducible nitricoxidesynthase,iNOS)3种类型。nNOS和eNOS合称为组成型(或原生型或固有型)一氧化氮合酶(constitutive nitricoxidesynthase,cNOS)。cNOS的调控机制与Ca2+浓度密切相关。诱导型NOS(iNOS)代表钙非依赖性异构体,iNOS仅被脂多糖(LPS)、白细胞介素1(IL-1)和α肿瘤坏死因子(TNF-α)[6]等细胞因子激活。NO也可以独立于NOS在低氧和/或酸性条件下自发发生亚硝酸盐还原作用,或由黄嘌呤氧化酶和细胞色素氧化酶c等不同酶的作用介导而形成。通过其他非酶途径产生的NO,例如皮肤组织、食物和肠道菌来源的NO衍生物亚硝酸盐等经体内外因素作用后转化为NO加入机体NO储备池,可以起到与酶促反应产生NO类似的调控作用[7]。
NO在机体内多组织脏器内发挥重要的生物效应,现阶段研究发现NO限速酶NOS在多种效应细胞中存在,并由特定通路激发作用。iNOS来源的NO经NF-κB-iNOS-NO通路,NF-κBp50和p65转移到核,参与机体炎症以及与细胞增殖、凋亡、衰老等细胞过程相关的多种基因转录[8]。在机体免疫系统中,NF-κB代表的转录因子家族成员调节iNOS基因:iNOS催化NO的产生,NO是在炎症和免疫应答中起作用的分子[9]。高浓度的NO具有普遍的免疫抑制作用,作为信号分子主要调节T细胞、B细胞的分化与效应功能,其主要供应途径分别是:①T细胞内eNOS、iNOS的表达产生内源性NO;②来自于微环境中树突细胞、巨噬细胞表达的外源性NO。此外,MAPK(ERK1/2,JNK1/2和p38)-iNOS-NO通路产生的NO也参与到炎症反应之中。eNOS来源的NO基于PI3K/AKT/eNOS/NO通路[10]、Ca2+/CaMKK或CaMKⅡ/AMPK/eNOS/NO通路、AMPK/AKT/eNOS/NO通路[11]等路径作用于内皮细胞影响脑缺血后适应、血压升降、血管生成、炎症变化和内皮细胞损伤程度。
3 卵母细胞成熟过程
卵母细胞(oocyte)是雌性动物独有的细胞,具有发育为完整生命个体的潜能。卵母细胞源于卵原细胞,其成熟过程可大体分为3个阶段,分别为LH激增触发减数分裂恢复引起生发泡破裂(GVBD)、经过快速分裂发育阶段停滞于第2次减数分裂中期(MⅡ)、受精作用诱导减数分裂完成,这期间需要多种激素和调节因子的共同参与。对人类女性而言,早在胚胎时期,女婴一对卵巢中的卵原细胞就已陆续分裂分化并产生约两百万枚初级卵母细胞,它们继续发育直到第1次减数分裂(meiosis Ⅰ)前期(prophase),并停滞于双线期(diplotene)阶段。在双线期停滞期间,卵母细胞虽细胞核发育受阻,但仍在积累各种营养物质,储存胚胎早期发育所需各类信息,使细胞体积不断增大。双线期阻滞持续时间一般较长,不同物种持续时间长短变化很大。人类卵母细胞的双线期最短可持续约15年,即女性发育至青春期并出现月经;最长可持续40~50年,即女性进入绝经期。当人类进入青春期时,约有40万个初级卵母细胞被保留下来,并在排卵前促性腺激素释放激素(gonadotropin-releasing hormone,GnRH)、促黄体生成素(LH)等生物因子的刺激下解除双线期阻滞恢复减数分裂,进入快速成熟期。第一次减数分裂恢复时,卵母细胞细胞核膨大、核仁增大且合成活跃,称为生发泡(germinal vesicle,GV),随后生发泡有组织地解体破裂,这标着卵母细胞成熟相关的减数分裂恢复。此时,卵细胞由GV期(germinal vesicle)进入生发泡破裂期(germinal vesicle breakdown,GVBD),初级卵母细胞中染色体浓缩、纺锤体形成,发育为次级卵母细胞并排出第1极体。此后,卵母细胞进入第2次减数分裂且再次停滞于中期Ⅱ(MⅡ)。排卵后,处于MⅡ停滞期的次级卵母细胞进入输卵管,等待受精,若完成受精则重新恢复减数分裂,形成一个有功能活性的成熟卵细胞和2个或3个无功能的极体,受精产生的受精卵发育为早期胚胎直至形成新生命;若次级卵母细胞未受精则进入多信号分子调控的衰老过程。
在卵母细胞成熟过程中,大多数初级卵母细胞尚未成熟就已退化。以家猪为例,其卵巢中存有卵原细胞110 000枚,但一生仅排卵400~2 000枚,一个发情周的排卵数为12~24枚,经外激素刺激可增加排卵数达30~40枚。如何利用这些珍贵的卵母细胞并合理激发其生殖潜能成为了近年来的热门议题。在人类辅助生殖方面,冷冻卵母细胞和胚胎技术可为错过最佳生育年龄或者患有肿瘤[12]疾病的女性提供生育机会。在家畜育种方面,卵母细胞及胚胎冷冻技术可与超数排卵、人工授精、胚胎移植等繁殖技术结合进而提高优良母畜的繁殖能力[13]。由于冻卵技术相对于精子冷冻技术不甚成熟,且卵母细胞冷冻及复苏过程中影响因素较多。因此,卵母细胞体外培养及长期保存方面需要探寻更为可靠有效的新途径。
4 NO调节的卵母细胞减数分裂成熟
4.1 NO调节的卵母细胞减数分裂停滞和GVBD
在哺乳动物卵母细胞中,细胞内的减数分裂停滞及物质变化由多种生物因子调控,其中高水平的cAMP起着主导作用。在体外培养条件下,从有腔卵泡内分离的卵母细胞中cAMP水平的稳定下降与减数分裂恢复同时发生,并且这种卵母细胞的自发成熟可以通过cAMP类似物或cAMP磷酸二酯酶(PDE)抑制剂来阻止[14]。近年来大量研究表明,卵母细胞停滞在前期Ⅰ所需的高水平cAMP分别通过G蛋白偶联受体(GPRs)通路和颗粒细胞间隙连接分别在卵母细胞中自主合成和胞间扩散来维持。在有腔卵泡中,卵泡微环境中的颗粒细胞产生高水平的cAMP,并通过间隙连接扩散到卵母细胞中。对于卵泡中的卵母细胞而言,细胞膜表面的跨膜蛋白激活型受体GPR3/GPR12受生殖激素的影响提高激活型G蛋白(Gs)活性,Gs活化腺苷酸环化酶(AC)催化ATP产生高水平的cAMP。cAMP在细胞内产生效应的方式是它能够将cAMP依赖性蛋白激酶(PKA)激活,PKA在A激酶锚定蛋白(AKAP)的调节下定位于下游促成熟因子(MPF)的抑制蛋白(如Wee1/Myt1)的激酶附近并提高其活性,使催化亚基周期蛋白依赖性蛋白激酶(CDK1)磷酸化,将MPF维持在稳定的非活性状态,导致双线期减数分裂停滞。MPF是存在于卵母细胞细胞质中的重要调节因子,由催化亚基周期蛋白依赖性蛋白激酶Cdk1(cdc2)和调节亚基细胞周期蛋白Cyclin B1组成。MPF处于稳定状态时,Wee1/Myt1使Cdk1的Thr14和Tyr15磷酸化,Cdk1与Cyclin B1结合为一无活性复合物,即前体MPF,维持卵母细胞双线期和减数分裂中期停滞。此外,PKA(以及其他未知因素)会使双特异性磷酸酶cdc25磷酸化且与胞质内蛋白螯合,从而阻止其发挥空间定位调节和使Cdk1的Thr14与Tyr15去磷酸化从而激活MPF的功能。其中,cdc25有3种亚型(A、B和C),它们都在小鼠卵母细胞中表达,但cdc25B对于减数分裂的恢复是不可或缺的,对于卵母细胞停滞于第1次减数分裂不能恢复的cdc25B基因敲除小鼠而言,可通过注射cdc25B mRNA来逆转该情况[15]。上述维持减数分裂的GPR3-Gs蛋白依赖性机制仅在有腔卵泡环境下进行,在GPR3基因敲除小鼠中,腔前卵泡中的卵母细胞在前期Ⅰ阶段仍然停滞,但在卵泡腔形成时自发恢复减数分裂。
研究表明,一氧化氮合酶eNOS、iNOS在雌性哺乳动物胚胎各个时期的卵巢表面上皮细胞、卵原细胞、卵母细胞、固缩坏死的卵原细胞和卵母细胞及血管内皮细胞都有表达,根据NOS的分布和NO作用广泛的特性,推测NO作为新型的胞间/内信使和一种不典型的递质参与了卵泡发育、黄体化、卵泡闭锁、排卵等过程,包括作用卵母细胞减数分裂成熟[16-17]。同时,由于维持卵母细胞减数分裂停滞的GPR3-Gs蛋白依赖性机制运行所需的限制性条件在临床研究中不便达成,为满足对早期腔前卵泡和离体卵母细胞的科研培养要求,在培养液中添加一氧化氮或一氧化氮供体将有利于维持卵母细胞双线期停滞并减少其提前衰老所带来的损失。NO是改变卵母细胞生理的主要信号分子之一[18],一氧化氮合酶(NOS)途径是卵周颗粒细胞和卵母细胞产生大量NO的主要途径,作为NO合成的主要限速酶,细胞中NOS浓度及分布可线性反映NO浓度分布情况。研究发现,在哺乳动物原始卵泡发育至有腔卵泡的过程中,eNOS和iNOS多数在颗粒细胞、卵母细胞膜层、卵母细胞和附近血管中存在,卵母细胞通过NOS介导途径产生的NO,足以维持哺乳动物个体从出生到排卵前卵泡微环境内的卵母细胞减数分裂停滞[19]。在双线期阻滞阶段,卵泡微环境中经由酶途径产生的NO利用其高脂溶性通过间隙连接由颗粒细胞向卵母细胞转移,或由卵母细胞通过NOS催化底物产生,维持卵母细胞内的高水平NO。在小鼠卵泡中,颗粒细胞内利钠肽前体C型(NPPC)与之利钠肽受体2(NPR2)相结合,刺激环磷酸鸟苷(cGMP)合成并通过旁分泌方式进入到卵母细胞之中;同时,NO与胞内铁离子一起激活作为胞内受体的颗粒状鸟苷酸环化酶(GC)催化GTP产生cGMP。胞内cGMP作为第二信使结合并抑制cAMP灭活激酶磷酸二酯酶3A (PDE3A)活性,减缓胞内cAMP降解并保持其高浓度水平,提高GPR3-Gs蛋白依赖性机制下游效应蛋白PKA活性,促进细胞周期因子25B(Cdc25B)磷酸化而抑制其活性,最终实现促成素因子(MPF)长期稳定。
哺乳动物生殖系统受到月经周期或发情期的调节,卵泡接受到的黄体生成素(LH)水平激增,LH刺激表皮生长因子(EGF)蛋白诱导的多种信号通路中断,导致间隙连接和类固醇生成通路破坏,使得卵丘卵母细胞复合体(COCs)细胞间通讯中断;NO、cGMP和cAMP等在内的多种信号分子向卵母细胞的传输中断。NO水平下降主要通过减少对cAMP降解酶PDE3A活性抑制的方式参与到胞内cAMP水平降低以恢复减数分裂的过程中。在LH/hCG诱导减数分裂从双线期停滞和生发泡破裂恢复的复杂过程中,涉及3种类型的NOS,特别是iNOS的在颗粒细胞和卵母细胞内表达受抑制明显。由于iNOS等途径产生的NO水平下降导致卵母细胞产出cGMP减少,同时LH可去磷酸化NPPC使得NPR2失活导致颗粒细胞中cGMP产出减少,卵母细胞中cGMP水平降低解除了对PDE3A活性的抑制而促进其对cAMP的降解。由于维持减数分裂停滞的关键因子cAMP水平下降,则PKA失活并与AKAPs亚基结合使其空间定位改变远离作用位点,解除对cdc25活性的抑制。cdc25B磷酸酶去磷酸化而活性增加,有效促使MPF亚基CDK1的Thr14和Tyr15去磷酸化,激活CDK1,成熟促进因子(MPF)双亚基结合状态破坏导致MPF去稳定化,并在Cdc25B的微调控制作用的影响下移入细胞核内诱导生发泡破裂(GVBD),哺乳动物卵母细胞退出双线期停滞状态进入减数分裂期[19-20]。上述研究表明,细胞内NO水平的降低可能诱导卵泡卵母细胞从双线期停滞恢复减数分裂。在LH/hCG激增或卵母细胞从其卵泡微环境中脱离的体外培养条件下,NOS活性降低,卵母细胞内NO水平降低,经由cGMP/PDE/Cdc25B/MPF途径介导的依赖于PKA失活的cdc25B的正确核迁移对于核MPF的激活和随后的GVBD发生至关重要。
体外培养条件下,NO对卵母细胞的调节具有双相性且取决于它的浓度。Anim等研究发现,较低浓度下(0.01 mmol/L)的一氧化氮供体如硝普钠(SNP)可诱导LH诱导的缝隙连接破坏、卵丘细胞扩张以及丝裂原活化蛋白激酶(MAPK)活性,实现减数分裂恢复;而较高浓度(0.5 mmol/L)却抑制体外培养的牛卵母细胞从双线期停滞恢复减数分裂[21]。在科研领域,NO供体成功已运用于防止小鼠、大鼠、猪、犬、牛等动物卵母细胞从双线期停滞期提早恢复减数分裂。虽然一氧化氮在卵泡卵母细胞二倍体停滞减数分裂恢复中的作用仍有争议,但NO在细胞中的双相作用已有大量报道,基于一氧化氮供体和iNOS抑制剂诱导减数分裂恢复的研究,我们提出iNOS途径介导的NO水平降低可能在哺乳动物排卵前卵母细胞减数分裂退出双线期停滞中起重要作用的结论。关于临床上NO双相性的运用,因目前仍未归整出准确的NO添加浓度与生物效应间的函数关系,而且此结果可能受到卵母细胞所处的时间、环境、周期以及所属物种等多种因素影响,需要更多的研究来阐明NO调节哺乳动物卵母细胞减数分裂细胞周期的分子机制,实现NO在卵母细胞培养中的生产运用价值。
4.2 NO参与的第2次减数分裂中期停滞(MⅡ停滞)
在多数哺乳动物发育过程中,继GVBD之后,进入核内的MPF激活mos,mos是负责有丝分裂原活化蛋白激酶(MAPK)途径的上游蛋白,MAPK使许多转录因子活化完成翻译,卵母细胞快速通过MⅠ期,作短暂停留或不停留,在排卵前或排卵时进入以第1极体排出为基本形态学特征的MⅡ阶段,经排卵进入输卵管并运行至输卵管壶腹部等待受精。家畜经超速排卵处理后,从输卵管中冲取的卵母细胞多处于该阶段。若错过受精窗口期,卵母细胞则经由细胞衰老介导的MⅡ停滞自发退出而走向凋亡。在哺乳动物(啮齿动物、猪和牛)中,MPF的作用因发育周期变化而改变,其中MPF活性在GVBD时增加,在MⅠ完成时下降,在MⅡ再次达到高活性水平,并继续升高直至受精[22]。其中MⅠ完成期间,MPF活性的下降不是由于CDK1的再磷酸化,而是由于细胞周期蛋白B1的降解,引起细胞周期蛋白B1降解的多泛素化是由一种称为后期促进复合物/环体(APC/C)的多亚基E3连接酶复合物控制的[23]。在MⅠ-MⅡ转换期间和MⅡ阻滞时,卵母细胞通过抑制APC活性的mos-MAPK-p90rsk-Emi途径重新建立MPF活性,防止细胞周期蛋白B1降解并稳定MPF,并在细胞抑制因子(CSF)的作用下导致MⅡ阻滞。
在排卵前后,哺乳动物下丘脑、垂体前部及后部均存在cNOS,推测NO可能通过调节GnRH的分泌来调节LH的分泌,也可能通过这一机制来影响卵巢类固醇的生成并刺激前列腺素的表达以促进卵泡破裂。由于LH激增抑制iNOS表达,COCs间隙连接中断,随之而来的NO下降导致卵泡内cGMP水平降低,这允许了MAP2K1和MAPK的顺序激活。MOS/MEK/MAPK信号通路通过蛋白激酶磷酸化的级联反应调节细胞周期,对维持卵母细胞的MⅡ期阻滞至关重要[24]。诱导原癌基因c-MOS的产物MOS诱导MEK磷酸化并提高其活性,进一步激活MAPK,MAPK与早期有丝分裂抑制剂2(Emi2)发生磷酸化作用抑制后期促进复合物/环体(APC/C)活性,APC/C活性降低可维持MPF稳定。或因卵母细胞衰老,MAPK活性耗竭会降低cAMP和Emi2水平,通过降低细胞周期蛋白依赖性激酶1(Cdk1)的特异性磷酸化和诱导细胞周期蛋白B1的降解来破坏MPF的稳定性,不稳定的MPF驱动哺乳动物卵母细胞自发退出MⅡ停滞。但关于MAPK3/1的耗竭原因以及下游调控机制与MPF的相互作用尚不明确,需要进一步实验研究。
MAPK激活的确切时间在各种哺乳动物中仍存在争议。虽然人们普遍认为MAPK途径对卵母细胞成熟至关重要,但MAPK激活和GVBD的时间顺序仍不清楚。在山羊中,MAPK在GV阶段以非活性形式存在,并在GVBD之后被激活;而在牛和马卵母细胞中,MAPK在GVBD之前被激活[25]。在啮齿动物中,MAPK激活发生在GVBD之后[26],并在涉及MAPK抑制剂和mos基因敲除小鼠模型试验中表明,在没有MAPK途径参与的情况下卵母细胞可以恢复减数分裂和第1极体的排出,但这些卵母细胞在MⅡ期停滞的能力受损。有报告称,MAPK3/1活性在猪和牛卵母细胞的MⅠ和MⅡ阻滞过程中达到最大值[27],Tongetal等也报告了猪卵母细胞在MⅡ停滞时表达高水平MAPK3/1活性。因此,至少在农场动物中,认为MAPK激活可能不是恢复双线期停滞减数分裂所必需的,而是MⅡ阻滞所必需的,这说明MAPK3/1通过Emi2磷酸化途径在维持MPF稳定中发挥重要作用。
除MOS/MEK/MAPK信号通路影响的卵母细胞MⅡ停滞及恢复途径外,NO参与的cGMP/PDE/Cdc25B/MPF途径也与MⅡ停滞的恢复相关。猪卵母细胞在恢复减数分裂期间eNOS表达增加的研究证明,卵泡卵母细胞能够产生足以调节自身生理效应的NO[28]。因此,虽然来自颗粒细胞的NO、cGMP和cAMP的供给因卵丘卵母细胞复合体(COCs)间隙连接破坏而中断,但是卵母细胞仍能产生足够的NO通过cGMP/cAMP/PKA/Cdc25B介导的途径调节Cdk1的特异性磷酸化及其活性,导致MPF积聚,阻止MPF去稳定化而维持MⅡ停滞;此外,经由MAPK(ERK1/2,JNK1/2和p38)-iNOS-NO通路产生的NO也参与到卵母细胞周期调节中。排卵后卵母细胞进一步衰老导致NOS的表达下降,使得卵母细胞中的NO水平不足以维持调节Cdk1特异性磷酸化及其活性的能力,导致稳定的MPF亚基游离分散,卵母细胞从减数分裂MⅡ停滞中恢复。
近年的研究也报道了NO在卵母细胞MⅡ期减数分裂细胞周期调节过程中的双相作用,表明NO可能在不同浓度基础上发挥保护作用或诱导卵泡凋亡的作用。NO供体如S-亚硝基、N-乙酰青霉胺(SNAP)以浓度依赖性方式防止小鼠和大鼠老年卵母细胞从MⅡ停滞中退出恢复减数分裂。低浓度NO诱导卵母细胞减数分裂恢复并在有腔卵泡的颗粒细胞中经Fas-FasL系统介导刺激细胞凋亡[29];高浓度则维持牛、小鼠和大鼠卵母细胞的MⅡ期停滞。现阶段实验室已使用NO供体防止小鼠和大鼠卵母细胞中MⅡ阻滞自发退出,延缓卵母细胞衰老,维持卵子质量和发育潜力,预防染色体异常[30]。其他报告显示,将刚排卵的卵母细胞暴露于NO供体条件下可防止透明带溶解时间缩短,降低自发性皮质颗粒丢失和纺锤体异常率[33]。体外实验研究表明,NO在大鼠[31]、牛[32]和人[33]模型中抑制颗粒/黄体细胞中雌二醇/孕酮的产生,而NOS抑制剂增强颗粒/黄体细胞的类固醇合成。同时,NO可能是一种抗氧化剂,能够减少卵母细胞中的活性氧(ROS)和过氧脂质自由基含量,NO的人工增加有助于恢复生物学上可利用的NO与导致衰老的自由基的过量产生之间的平衡。上述研究结果支持了一定浓度的NO具有维持卵细胞的质量、受精能力和延缓衰老的观点。因此,从细胞外来源补充一氧化氮将有利于防止排卵后卵子衰老介导的M-Ⅱ停滞的自发退出以及卵子质量的恶化。
5 结论
在现代生物研究和临床医疗中,我们重点关注卵母细胞的发育潜能和卵母细胞抗衰老以获得更长的等待受精时间等问题。参考iNOS在哺乳动物排卵前后的强烈表达的研究结果以及小鼠注射iNOS抑制剂,如氨基胍(aminoguanidine,AG),并敲除eNOS基因后,其卵母细胞均不能正常发育的试验结果,认为试验中可运用iNOS抑制剂如氨基胍(AG)降低酶途径产生的NO水平,诱导从卵巢卵泡中直接抽取的卵母细胞从双线期停滞中恢复,及时达到后续试验或辅助生殖所要求状态。在卵母细胞抗衰老问题的研究方面,有报道称NO可能是一种抗氧化剂,能够减少卵母细胞中的活性氧(ROS)和过氧脂质自由基含量,并通过抑制颗粒细胞凋亡阻止卵泡闭锁。因此,在卵母细胞体外培养时,人工增加NO有助于恢复细胞在生物学上可利用的NO与导致衰老的自由基的过量产生之间的平衡。NO参与防止排卵后衰老介导的MⅡ停滞自发退出的一个可能机制是通过激活鸟苷酸环化酶(GC),使得cGMP的产生增加;另一方面,细胞内NO水平的高低与细胞内cGMP、cAMP、cdc25B水平的变化相关联。因此,充分利用NO在卵母细胞成熟过程中双相作用,维持MPF稳定以阻止排卵后衰老介导的MⅡ阻滞的自发退出;在体外培养条件下,细胞外来源人工补充NO或利用NO供体将有利于防止排卵后卵母细胞衰老介导的MⅡ停滞的自发退出和卵母细胞质量的恶化。
卵母细胞从双线期停滞阶段恢复减数分裂直到MⅡ停滞阶段的发育过程是关乎其细胞质量的重要阶段,经由NOS通路产生的NO作为重要信号分子参与调节卵母细胞减数分裂发育成熟的过程。尽管NO在减数分裂细胞周期中的调节作用在不同物种中仍存在争议,但基于现有研究,合理运用NO的双相作用调节卵母细胞发育,作为延长卵母细胞寿命、增加其等待受精时间的可行办法已有一定的试验基础。但是在不同物种及细胞状态下的NO、NO供体或NOS抑制剂的参考用量及用法尚未明确,还需要更多的试验证据加以总结,进一步阐明NO在卵母细胞发生过程中的调节机制,深入研究NO浓度、NO供体以及NOS抑制剂等在生产实践中的运用条件将有助于深刻理解卵母细胞成熟这一重要的生理过程,可为调控动物生殖和治疗生殖疾病提供理论基础,也为人类的生殖健康维护和生产、医学临床应用提供重要的参考。
