基于聚集诱导发光的计算化学实验设计
2024-05-17魏玉玲
魏玉玲
(南京邮电大学材料科学与工程学院,南京 210023)
0 引 言
聚集诱导发光(AIE)[1-3]在溶液中不发光或者发光很弱,而在固态或聚集状态下发光增强。在材料制备实验课中学生可以观察到明显的实验现象,但对其发光机理却知之甚少,学生期望能进一步探索研究其发光机理。本实验选取5,5′-双(2,2-二苯基乙烯基)联噻吩(BDPV2T)[4]为研究对象,其主链是具有π 共轭结构的噻吩苯环结构,两端连有AIE 特征基团,分子结构简单计算量小,适合开展实验教学。
本文以AIE 材料的分子运动受限机理(RIM)为例,设计一个计算化学实验,为材料类专业本科生提供一个认识和学习计算化学的方式。提升学生在光电功能材料设计、开发方面的能力,加深学生对相关知识和基本理论的理解,掌握计算化学基本方法并迁移至不同场景应用。
1 计算理论与方法
1.1 计算软件
本课程所用计算软件主要有Gaussian、MOMAP,其中几何结构优化和频率计算使用Gaussian 进行[5],速率计算和正则模式分解使用MOMAP软件[6]计算。
Gaussian软件是目前应用最广泛的一款计算化学软件,能够适配不同规格的计算级别。可以执行结构优化、振动分析、吸收发射光谱等一系列计算任务。
MOMAP(分子材料性质预测软件)是由清华大学帅志刚教授课题组、中国科学院化学研究所有机固体实验室理论组和鸿之微科技(上海)股份有限公司联合研发的一套研究多原子分子光谱、辐射和无辐射过程的软件包。该软件操作简便,导入相应软件后,修改相应.inp文件即可计算,适合本科生和研究生教学。
两个软件的配合使用不仅可以使学生锻炼实际操作能力,更有助于学生从理论层面理解化学现象,加深知识的理解和应用,为学生产生知识的迁移提供帮助。
1.2 密度泛函和含时密度泛函理论
密度泛函理论(DFT)提供了一个精确的理论框架,基态能量以电子密度作为变分函数来求解,通过计算多体系波函数产生可直接观测的量。Hohenberg等[7]证明了静态多电子系统的所有可观测性质可以从单体基态密度中提取,为DFT 提供了坚实的理论依据。
含时密度泛函理论(TD-DFT)是对多体系非相对论时间相关量子力学的一种严格重构,中心定理是Runge-Gross定理。TD-DFT可以实现计算负荷与精度的良好平衡,因此成为目前最准确和可靠的预测固体物理、化学和生物物理学激发态性质的工具之一[8]。
目前绝大多数量化软件都支持含时密度泛函理论,而由于TD-DFT 计算精度受泛函、基组、环境等影响,因此实际计算过程中需要根据实际情况选择合适的计算级别。
1.3 溶剂模型与ONIOM模型
考虑到环境对分子的耦合作用,本工作分别采用PCM溶剂模型和ONIOM模型中的QM/MM方法来模拟分子在溶液和晶体中的环境影响。
隐式溶剂模型把溶剂环境当作具有固定介电常数的连续介质[9],目前应用最广的就是极化连续溶剂模型(PCM),可以使用Gaussian 中的SCRF 关键词来实现[10-11]。
在研究聚集体激发态过程中,单纯使用量子力学(QM)或者分子力学(MM)都不能够满足需求。量子力学/分子力学组合(QM/MM)方法是Warshel 等[12]在20 世纪70 年代提出,两者因此在2013 年获得诺贝尔奖[13]。本文使用Gaussian 程序中的ONIOM 模型[14],在QM程序中引入MM的计算。
1.4 激发态衰减理论
图1 为简化的雅布隆斯基能级图,几乎包括分子的所有基本光物理过程。光吸收过程用橙色的直线箭头表示,基态到第一单重激发态的吸收跃迁为S0 →S1。绿色直箭头表示荧光,为释放光子的辐射过程。荧光过程对应图中的S1→S0,根据跃迁规则,单重态到单重态的多重度不发生改变,跃迁允许,因此辐射速率kr一般比较大((107~109)s-1)。
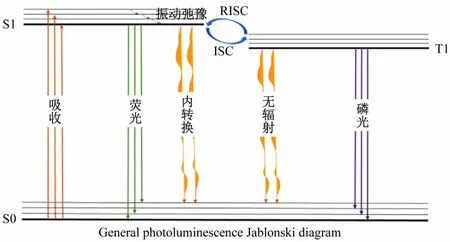
图1 光致发光的雅布隆斯基能级图(粗水平线表示各个电子态能量的最小值,水平细线表示振动能级)
无辐射跃迁过程包括无辐射耗散、系间窜越(ISC)和反系间窜越(RISC)。其中无辐射过程是指分子通过无辐射驰豫方式耗散能量,对应图中的内转换、无辐射和电子态内的振动弛豫过程。根据激发态衰减理论,辐射过程与无辐射过程的竞争决定了一个分子是否发光。
以第一单重激发态的衰减过程为例,荧光辐射速率kr、S1-S0的内转换速率kIC、S1~T1的系间窜越速率(kISC),3 个过程互相竞争。对于荧光分子,由于π-π*跃迁的自旋轨道耦合非常小,可以忽略kISC,因此荧光量子产率就可以表示为辐射速率与能量衰减总速率的比值[15]:
1.5 辐射速率与无辐射速率计算
为使计算结果更加精确,本文使用MOMAP 软件计算激发态衰减速率。
(1)荧光速率。辐射速率计算公式根据爱因斯坦自发辐射方程得到:
其中:Evert为垂直激发能;f为振子强度。考虑电子态与振动多能级的耦合和温度效应kr的单位是s-1。
(2)内转换速率。对于无辐射衰减,应用二阶微扰近似,将较小的二阶项∂2Φi/∂忽略后,利用Condon近似并对δ函数进行傅里叶变换后,得到kIC的表达式[16-17]:
2 结果处理与讨论
2.1 分子结构分析
根据电子结构理论,激发态结构变化影响电子分布。为了讨论溶液状态和晶体状态对分子结构的影响,分别使用PCM 溶剂模型和ONIOM 模型优化了基态S0和第一单重激发态S1的结构,并定义了基态-激发态的结构变化最大的4 个二面角(见图2)。
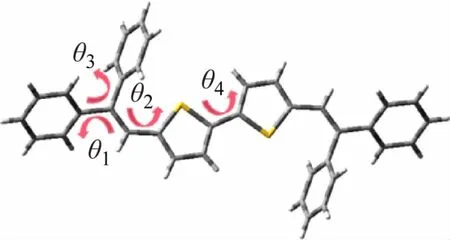
图2 分子结构及二面角定义
不论是在溶液中还是晶体模型中,对比优化后的分子结构,发现基态到激发态的结构驰豫最大的部分均为二面角θ3(ΔS1-S0=20.2°)。但比较溶液和晶体,在晶体中θ3的变化只有3.96°(ΔS1-S0=3.96°)。很明显二面角的转动在晶体模型中被显著抑制(见表1)。
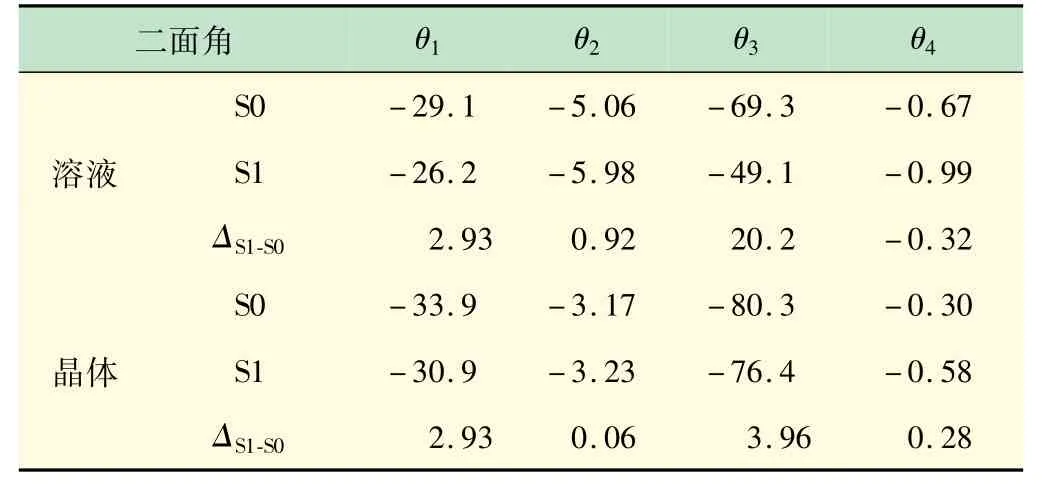
表1 在溶液和晶体中优化后二面角(θ1 ~θ4)的变化
2.2 吸收光谱分析
考虑到聚集诱导发光分子的结构具有柔性,也为了进一步验证计算结果的可靠性,把使用ONIOM模型计算得到的吸收发射波长(见图3)与实验测得的数据λabs(450 nm)、λemi(533 nm)做对比。
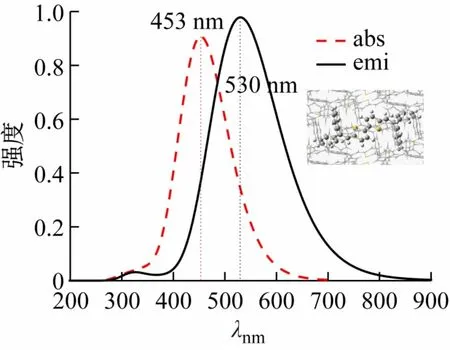
图3 采用ONIOM模型计算得到的吸收发射波长
计算得到的吸收发射波长与实验测得的吸收发射波长相差仅有3 nm,在误差允许范围内。计算数据与实验数据有良好的对应,表明选用ONIOM模型和计算结果的可靠性。
2.3 波函数分析
根据电子结构理论,结构变化影响电子云分布。在图4 中分别展示了用PCM溶剂模型与ONIOM溶剂模型计算得到的HOMO、LUMO轨道能量、电子云分布及其能级差、振子强度。电子激发态显示出明显的ππ*特征,与溶液中相比,在晶体中分子的电子云在侧链苯环上几乎没有分布,表明整体的电子云分布更加局域,HOMO-LUMO 之间的能级差增大,发射波长蓝移。
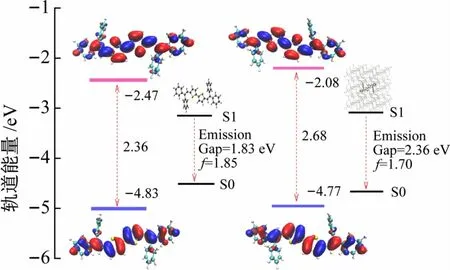
图4 溶液(左)和晶体(右)中的HOMO-LUMO电子云分布及垂直发射能和振子强度
2.4 正则模式分析
无辐射过程是指分子通过无辐射驰豫方式耗散能量,对应图中的内转换、无辐射和电子态内的振动弛豫过程。根据激发态衰减理论,辐射过程与无辐射过程的竞争决定了一个分子是否发光。
本文讨论影响无辐射衰减的两个主要因素,分别是黄-昆(HR)因子和重组能。图5 显示振动对HR因子的影响主要集中在低频区(0 ~100 cm-1),溶液中最高的贡献来自频率为29.93 cm-1的中心噻吩环的转动(HR=10.86),而在晶体中这一振动被抑制(HR=0.66),HR因子降低了93.92%。
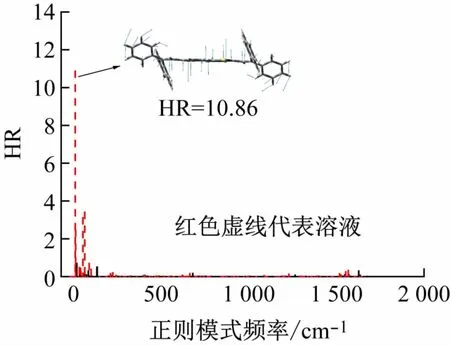
图5 HR因子对正则模式频率的分解
对重组能的影响主要集中在高频(1 500 ~1 750 cm-1)和低频区(0 ~250 cm-1)。溶液中高频区贡献主要来自1 557.68 cm-1的C—H键角的剪切振动,在晶体中这种振动模式分别被抑制,两者的重组能从溶液中502.05 cm-1减小至晶体中的154.30 cm-1降低了3.3 倍(见图6)。低频区贡献主要来自29.93 cm-1的中央噻吩环的转动(RE =325.2 cm-1)以及83.16 cm-1侧链苯环的转动(RE =282.57 cm-1),在晶体中这两种振动模式对重组能的贡献分别为25.26 和9.17 cm-1,即,从溶液到晶体,这两种振动模式的重组能分别降低了92.23%和96.75%。在PCM溶剂模型中计算得到的总重组能为683 meV,而在ONIOM晶体模型中计算得到的总重组能为403 meV,表明晶体环境可以有效降低体系重组能。
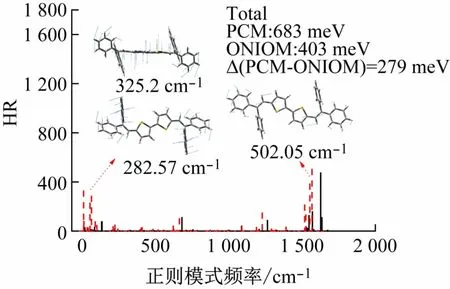
图6 重组能对正则模式频率的分解
总体看来,相比较于溶液环境,在晶体环境中重组能和HR 因子均有大幅的降低,总重组能降低了279 meV,降低了40.8%。
2.5 速率过程分析
基于基态激发态电子结构和振动频率,通过热振动关联函数方法计算了荧光辐射和无辐射的速率,见表2。结果表明,溶液和晶体中辐射速率相当但无辐射速率降低了4 个数量级,导致晶体中的荧光量子产率比溶液中增大了2 个数量级。为了评估计算结果的合理性,把影响辐射速率的因素振子强度、跃迁偶极矩、绝热能级差如表3 所示。
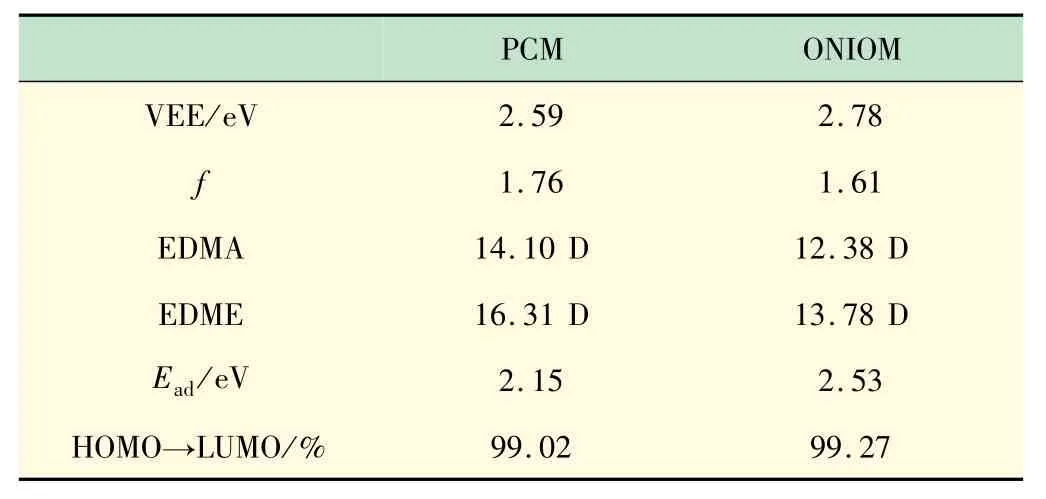
表2 垂直能级、振子强度、电跃迁偶极矩和S1 态轨道贡献
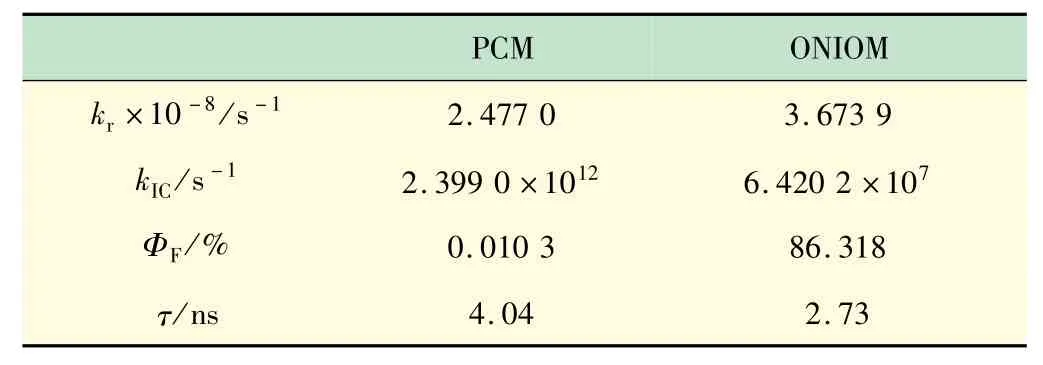
表3 计算得到的辐射速率、无辐射速率、荧光量子产率和荧光寿命
与溶液相比,晶体中的振子强度和跃迁偶极矩均降低,但绝热能级差增大,根据爱因斯坦自发辐射方程,辐射速率虽有增大,但不明显。晶体中的荧光量子产率比溶液高出接近3 个数量级,主要原因是晶体中分子的无辐射耗散被显著抑制。溶液中的无辐射速率是2.399 0 × 1012s-1,而在晶体中无辐射速率为6.420 2 ×107s-1,降低了接近5 个数量级,这与上节分析的重组能和HR因子的降低相印证。
综上所述,晶体环境可以有效抑制AIE 分子中转子的转动和部分键角的振动,减小HR因子和重组能,大大降低分子的无辐射耗散速率,从而实现从溶液到晶体的发光增强。
3 融合课程思政建设
聚集诱导发光理论作为中国科学家首次提出并逐步发展成熟的科学理论具有很强的思政教育意义。唐本忠院士在科学研究中提出了“Together We Shine,United We Soar!”的合作理念,团结了一批优秀顶尖人才,使得中国在聚集诱导发光领域处于全世界领先地位。郑州大学化学学院把AIE 理论作为课程思政元素引入本科教学,并取得了良好的效果[18]。本实验以一种AIE材料为例,从理论计算的角度出发设计了一个计算化学实验课,第一次把理论计算与AIE 结合起来用于实验教学。通过该实验课让学生初步理解AIE材料发光的理论机理,向学生传递“见人所未见,思人所未思”的科研理念,帮助学生树立远大理想,立志科研报国。
4 结 语
本文基于一种聚集诱导发光材料,以计算化学实验为抓手,对限制分子内振动机理进行了系统的探索。研究表明晶体环境能有效限制聚集诱导发光分子中转子的振动,降低无辐射耗散,提高荧光量子产率。本文以此设计了一个计算化学实验课,促进学生对聚集诱导发光的理论机理进行深入理解和掌握,充分调动学生学习的积极性,提升理论计算实验教学的魅力。该实验把理论和实践进行结合,增强了计算化学和材料学的交叉融合,有利于培养学生综合素质,同时拓宽学生科研视野、提升学生动手实践水平。
