A nanobody-based blocking enzyme-linked immunosorbent assay for detecting antibodies against pseudorabies virus glycoprotein E
2024-05-13HuanhuanPinpinJiSiyuLiuZiweiZhangLeiWangYaniSunBaoyuanLiuLizhenWangQinZhao
Huanhuan Lü ,Pinpin Ji ,Siyu Liu,Ziwei Zhang,Lei Wang,Yani Sun,Baoyuan Liu,Lizhen Wang ,Qin Zhao
Department of Preventive Veterinary Medicine,College of Veterinary Medicine,Northwest A&F University/Shaanxi Scientific Observing and Experimental Station of Veterinary Pharmacology and Diagnostic Technology,Ministry of Agriculture and Rural Affairs,Yangling 712100,China
Abstract Pseudorabies (PR) is an acute infectious disease of pigs caused by the PR virus (PRV) and results in great economic losses to the pig industry worldwide.PRV glycoprotein E (gE)-based enzyme-linked immunosorbent assay (ELISA) has been used to distinguish gE-deleted vaccine-immunized pigs from wild-type virus-infected pigs to eradicate PR in some countries.Nanobody has the advantages of small size and easy genetic engineering and has been a promising diagnostic reagent.However,there were few reports about developing nanobody-based ELISA for detecting anti-PRV-gE antibodies.In the present study,the recombinant PRV-gE was expressed with a bacterial system and used to immunize the Bactrian camel.Then,two nanobodies against PRV-gE were screened from the immunized camel by phage display technique.Subsequently,two nanobody-HRP fusion proteins were expressed with HEK293T cells.The PRV-gE-Nb36-HRP fusion protein was selected as the probe for developing the blocking ELISA (bELISA) to detect anti-PRV-gE antibodies.Through optimizing the conditions of bELISA,the amount of coated antigen was 200 ng per well,and dilutions of the fusion protein and tested pig sera were separately 1:320 and 1:5.The cut-off value of bELISA was 24.20%,and the sensitivity and specificity were 96.43 and 92.63%,respectively.By detecting 233 clinical pig sera with the developed bELISA and a commercial kit,the results showed that the coincidence rate of two assays was 93.99%.Additionallly,epitope mapping showed that PRV-gE-Nb36 recognized a conserved conformational epitope in different reference PRV strains.Simple,great stability and low-cost nanobody-based bELISA for detecting anti-PRV-gE antibodies were developed.The bELISA could be used for monitoring and eradicating PR.
Keywords: nanobody,nanobody-HRP,blocking ELISA,PRV-gE,antibody
1.lntroduction
Pseudorabies (PR),or Aujeszky’s disease,caused by the PR virus (PRV),is an acute infectious and fatal disease and brings huge economic losses to the pig industry worldwide (Lee and Wilson 1979;Liuet al.2021).This disease is endemic in pigs and causes various clinical symptoms,including neurological,respiratory,and reproductive disorders (Mettenleiter 2003;Linet al.2020).The PRV genome is a double-stranded linear DNA virus and encodes more than 70 proteins.The glycoprotein E (gE) is essential to viral pathogenicity,while it is not necessary for PRV replication (Sunet al.2022).Currently,some PRV-gE-deficient vaccine strains are widely used to control PRV infection in pig flocks because the absence of gE decreases the virulence of PRV (van Oirschotet al.1990).Therefore,an antibody against PRV-gE is a marker to distinguish vaccine-immunized pigs from wildtype virus-infected pigs (Anet al.2013).A fast,simple,responsive,and low-cost enzyme-linked immunosorbent assay (ELISA) to detect anti-PRV-gE antibodies in the pig sera is the first measure of PR eradication (Boonhamet al.2014;Liuet al.2019;Chenet al.2020).
Over the past decades,different types of ELISAs with polyclonal and monoclonal antibodies as reagents,including indirect and blocking ELISAs (bELISAs),have been developed to detect anti-PRV-gE antibodies in the pig sera (Aoet al.2003;Chenget al.2021;Panet al.2022).As we know,enzyme-conjugated antibodies are the essential reagents for developing sensitive,specific,and reproducible ELISA.However,conventional antibodies have some limitations,such as the required affinity purification,labels,and use of secondary antibodies.Therefore,although several commercial ELISA Kits for detecting anti-PRV-gE antibodies have been developed,these kits are relatively high costs and require lengthy production times (Guet al.2015;Liet al.2021).
Nanobodies are derived from the variable domains of Camelid heavy chain antibodies (VHHs) (Bannaset al.2017).Compared with the conventional antibodies,they have many attractive features,including small size (about 15 kDa,4 nm long and 2.5 nm wide),high stability,ease of genetic engineering,and thermal and chemical resistance (De Meyeret al.2014;Vogt 2016).Recently,nanobodies have been widely used to develop ELISAs to diagnose diseases based on their attractive features (Salvadoret al.2019;Yuet al.2021).Compared with traditional antibody-based ELISAs,these novel nanobodies-based ELISAs reduce testing time,simplify procedures and reduce costs because of the omission of labeling and incubation of secondary antibodies (Muyldermanset al.2009;Maet al.2019;Shenget al.2019).However,few reports about nanobody-based ELISAs for detecting anti-PRV-gE antibodies in pigs have been reported.In this study,the PRV-gE protein was expressed using theEscherichia colisystem and then used as an antigen to immunize Bactrian camels and to screen nanobodies (Fig.1-A).Afterward,the nanobodies with horse radish peroxidase (HRP) were produced using the platform to express the nanobody-HRP fusion protein in the HEK293T cells (Fig.1-B).Subsequently,a bELISA using the nanobody-HRP fusion probe was developed to detect antibodies against PRV-gE (Fig.1-C).The assay can distinguish gE-deficient vaccinated pigs from wildtype virus-infected pigs and showed good specificity and sensitivity.Importantly,the bELISA is unnecessary to use the enzyme-labeled second antibody because of the nanobody fused with HRP.The assay is an ideal method for eradicating PRV infection combined with using a PRVgE-deficient vaccine.
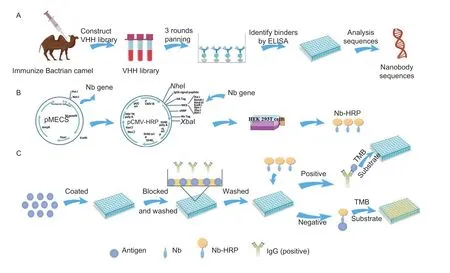
Fig.1 Schematic representation of screening nanobodies,production of the nanobody-horse radish peroxidase (Nb-HRP) fusion protein,and development of nanobody-based blocking ELISA (bELISA).A,screening nanobodies from an immunized camel by phage display technology.B,platform for expressing nanobody-HRP fusion proteins.C,designation of the developed bELISA.
2.Materials and methods
2.1.Cells,viruses,and vectors
HEK293T and PK-15 cell lines were purchased from American Type Culture Collection (ATCC) and cultured in Dulbecco’s Modified Eagle’s Medium (Life Technologies Corp,USA) containing 10% fetal bovine serum (FBS,Gibco,USA) at 37°C in 5% CO2.The PRV (strain Ea kindly provided by Prof.Xiao Shaobo in Huazhong Agricultural University,China,GenBank accession number KX423960.1) stocks were grown in the PK-15 cells and had the 10-6.5TCID50mL-1.The pMECS vector was kindly provided by Prof.Serge Muyldermans in Vrije Universiteit Brussel,Belgium and used to construct the library of VHH (Vinckeet al.2012).According to a previous description,the pCMV-N1-HRP vector with His-tag was constructed using the commercial pEGFP-N1 vector (Clontech,Japan) as the backbone (Shenget al.2019).
2.2.Serum samples
To determine the cut-off value of bELISA,a total of 108 sera from the specific-pathogen-free (SPF) pigs were used.For sensitivity and specificity,84 positive and 149 negative pig sera for anti-PRV-gE antibodies were separately tested using the bELISA.A commercial ELISA Kit (IDEXX,USA) was applied to confirm these positive and negative sera.The 233 clinical sera were collected from pig farms around Shaanxi,China.To determine the cross-reaction of the developed bELISA,32 sera from the pigs immunized with PRV gE-deficient vaccine and 71 ones positive for antibodies against other swine viruses,including porcine reproductive and respiratory syndrome virus (PRRSV) (n=24) confirmed by IDEXX PRRS X3 antibody ELISA Kit,porcine circovirus type 2 (PCV2) (n=15) confirmed by VDPro®PCV2 AB ELISA Kit (MEDIAN,Korea),transmissible gastroenteritis virus (TGEV) (n=20) confirmed by abbexa TGEV-Ab ELISA Kit (abbexa,UK) and swine hepatitis E virus (sHEV) (n=12) confirmed by Beijing WANTAI HEV Antibody ELISA Kit (WANTAI BioPharm,China),were tested using the assay.
2.3.Expression and purification of the recombinant PRV-gE
The main antigen coding region of the PRV-gE gene was synthesized and ligated into the commercial vector pET-28a by Azenta Life Sciences Company (Jiangsu,China).After sequenced,the positive plasmid was transformed intoE.coliTransetta (DE3) cells for expression (TransGen Biotech,Beijing,China).The positive recombinant bacteria were induced with 0.1 mmol L-1isopropyl-beta-D-thiogalactopyranoside (IPTG) and cultured for 24 h at 16°C.Then,the bacteria were collected by centrifugation at 10,000×g for 15 min at 4°C.The precipitation was resuspended in phosphate-buffered saline (0.1 mol L-1PBS,pH=7.4) and sonicated by the ultrasonic instrument.After that,the recombinant protein was purified by a cOmplete His-Tag Purification Resin based on the manual instructions (Roche,Germany).Finally,expression,purification,and antigenicity of the recombinant PRV-gE were analyzed by SDS-PAGE and Western blotting using the positive pig serum sample for anti-PRV antibodies as the primary antibody.
2.4.Camel immunization and library construction
A 4-year-old male Bactrian camel was immunized with the purified PRV-gE (1 mg mL-1) by the subcutaneous route five times as previously described (Liuet al.2015).For the first time,the protein (1 mg mL-1,1 mL) was emulsified with an equal volume of Freund’s complete adjuvant (Sigma,USA).Four additional immunizations were performed every 2 weeks with the same volume of Freund’s incomplete adjuvant.After the fifth immunization,the serum samples were collected from the immunized camel to evaluate the titers of anti-PRV-gE antibodies with indirect ELISA.Then,the peripheral blood lymphocytes (PBLs) were isolated from the fresh blood samples collected 7 d after the fifth immunization.Total RNA was extracted using RNeasy®Plus Mini RNA Extraction Kits (QIAGEN Bioinformatics,Germany) and used as the templates for cDNA synthesis using Reverse Transcription Kits (TransGen Biotech,Beijing,China).The VHH genes were amplified by the nested PCR with primer pairs CALL001 (5´-GTCCTGGCTGCTCTTCTACAAGG-3´),CALL002 (5´-GGTACGTGCTGTTGAACTGTTCC-3´),and VHH-FOR (5´-CAGGTGCAGCTGCAGGAGTCTGGGGGAGR-3´),VHH-REV (5´-CTAGTGCGGCCGCTGAGGAGACGGTGA CCTGGGT-3´) according to a previous description (Liuet al.2015).The nested PCR products were ligated into phagemid vector pMECS using thePstI andNotI (underlined in the primers) enzyme sites.The recombinant phagemids were electro-transformed into freshly competentE.coliTG1 cells,and the positive rate of the constructed library was determined by PCR amplification with primers MP57 (5´-TTATGCTTCCGGCTCGTATG-3´) and VHH-REV (Vinckeet al.2012).Finally,48 clones were randomly selected for sequencing to analyze the library’s diversity.
2.5.Screening and identification of specific nanobodies against PRV-gE
According to the previous descriptions,three rounds of bio-panning were performed to screen nanobodies using PRV-gE as coating antigens (Liuet al.2015).Briefly,the 96-well plate was coated with PRV-gE (100 μg mL-1) diluted in PBS (0.1 mol L-1,pH 7.2,100 μL perwell) overnight at 4°C.Then,the plates were blocked with 2.5% (w/v) nonfat dried milk in PBS (200 μL per well) at 37°C for 1 h and were washed four times with PBS’T (PBS with 0.05% Tween-20 (v/v)).Then,the rescued phage particles were prepared at a concentration of 4.4×1011pfu mL-1as in a previous study (Liuet al.2015) and incubated in the plates (100 μL per well) for 2 h at 25°C.After the plates were washed with PBS’T again,the phage particles were eluted with 100 mmol L-1trimethylamine (100 μL per well) (Sigma,USA) for 10 min at 25°C following to be neutralized with 1 mol L-1Tris-HCl (pH 7.4).The eluted phage particles infectedE.coliTG1,and the M13K07 helper phage was added to rescue the phage particles.The second and third rounds of biopanning were performed based on the above mentioned procedures.After three rounds of screening,the PRV-gEspecific phage particles were enriched and evaluated with polyclonal phage ELISA.Then,96 clones from the thirdround plates were randomly selected and cultured in liquid culture.The bacteria were collected by centrifugation and repeatedly freeze-thawed after being cultured overnight.By centrifugation again,the supernatants were the periplasmic extracts containing the nanobodies and tested for identifying the presence of specific nanobodies against the recombinant PRV-gE by indirect ELISA.Finally,all positive clones were sequenced and classified based on their complementary determining regions (CDRs) amino acid sequences.
2.6.Expression of nanobody-HRP fusion protein against PRV-gE
Based on the previous description,the nanobody-HRP fusion protein against PRV-gE was expressed in the HEK293T cells (Yamagata and Sanes 2018;Shenget al.2019).Briefly,the VHH genes encoding nanobodies were cloned into pCMV-N1-HRP with His-tag byPstI andNotI enzyme digestion following ligation with T4 enzymes.Then,the positive recombinant plasmids were confirmed by sequencing.The HEK293T cells were transfected with the positive plasmids using polyetherimide (PEI,Polysciences Inc.,Warrington,USA) agents for producing the nanobody-HRP fusion protein.After the cells were transfected for 3 d,the medium containing fusion proteins were harvested and filtered through 0.45-μmpore cellulose acetate membranes.Expression and titers of the fusion protein in the medium were separately identified by indirect immunofluorescence assay (IFA) and direct ELISA using the recombinant PRV-gE as the coated antigen and PEDV-N as the coated negative antigen.For IFA,after the medium was collected,the cells were fixed and incubated with anti-His monoclonal antibodies,followed by FITC-goat anti-mouse IgG antibodies (Jackson ImmunoResearch Laboratories,USA).The 96-well plates were coated with the purified recombinant PRV-gE (100 ng per well) for direct ELISA using PBS at 4°C overnight.Then,after washing three times,the plates were added into 100 μL of different medium dilutions and incubated for 1 h at room temperature (RT).After another three times’ washings,3,3´,5,5´-Tetramethylbenzidine (TMB) was added to the plates for a colorimetric reaction,and then,the OD450nmvalues were read using an automatic microplate reader (Bio-Rad,USA) after the reaction was stopped with 3 mol L-1H2SO4.The nanobody-HRP fusion protein with higher titer to react with PRV-gE was selected to develop the bELISA.
2.7.Establishment of the blocking ELlSA to detect anti-PRV-gE antibodies in pig sera
Using the nanobody-HRP fusion protein as the reagent,the bELISA was designed.First,the optimal amount of coating antigen and dilution of the medium containing the fusion protein were determined using a checkerboard titration with direct ELISA.Different amounts of the recombinant PRV-gE included 100,200,400,and 800 ng/well.The medium dilutions were 1:20,1:40,1:80,1:160,1:320,and 1:640.The optimized conditions were determined when the OD450nmvalues of direct ELISA were approximately 1.0.Second,the dilution of tested pig sera in the bELISA was optimized.Each four positive and negative pig sera for anti-PRV-gE antibodies was diluted with 1:5,1:10,1:20,1:40,1:80,and 1:160.And then,they were all tested with the bELISA using the optimal amount of coated antigen and dilution of the medium.The optimized dilution was determined when the smallest ratio of OD450nmvalues between the positive and negative serum (P/N) was obtained.Third,the incubation times of pig sera and nanobody-HRP fusion separately with the plate and the times of colorimetric reaction were also optimized.The incubation times of positive or negative pig sera with coated antigens were tested for 10,20,30,40,50,and 60 min.The incubation times of nanobody-HRP fusion with the plate after incubation with pig sera were tested for 10,20,30,40,50,and 60 min.The colorimetric reaction times were designed for 5,10,15,and 20 min.All three optimal times were selected as the smallest P/N ratios.
After the above conditions were optimized,the operations of the developed bELISA were as follows.The 96-well ELISA plates were coated with the optimized amount of recombinant PRV-gE and incubated overnight at 4°C.Then,the plates were blocked with blocking buffer (2% BSA in PBS) at 37°C for 1 h.After being washed with PBS’T three times,the wells were added with the optimal dilutions of testing pig sera in the blocking buffer (100 μL) and then incubated for optimized times at 37°C.After being washed three times with PBS’T again,the optimal dilutions of nanobody-HRP fusions in the blocking buffer (100 μL) were added and incubated for optimal times at 37°C.After being washed three times again,the 100 μL TMB was added into each well and colored for optimized times at RT.As a final step,the 3 mol L-1H2SO4(50 μL per well) was used to stop the colorimetric reaction,and the OD450nmvalues were read by an automated ELISA plate reader.The formula for calculating the blocking rate (PI) value is PI=(1-OD450nmvalue of positive sera/OD450nmvalue of negative sera)×100%.
2.8.Determination of the cut-off value for the developed blocking ELlSA
The 108 negative pig sera for anti-PRV antibodies were used to calculate the cut-off value of the bELISA.These sera were confirmed negative by a commercial ELISA Kit (IDEXX PRV/ADV gI Ab Test,IDEXX Laboratories,Inc.,Westbrook,ME,USA).Then,the cut-off value was calculated with the mean PI value (X) of the negative samples (N)+3×standard deviations (SDs).The PI values of testing serum samples greater than or equal to the cut-off value were considered positive for anti-PRV-gE antibodies.Conversely,the serum samples are negative.
2.9.Evaluation of specificity,sensitivity,reproducibility,and stability of the blocking ELlSA
The 84 positive pig sera for anti-PRV-gE antibodies were tested to determine the sensitivity of the developed bELISA,and different dilutions (from 1:5 to 1:2,560) of eight positive pig sera were tested to determine the detection limit.
To determine the specificity of bELISA,149 negative pig sera for anti-PRV-gE antibodies were tested.Meanwhile,the cross-blocking bELISA was evaluated using the pig sera from the pigs immunized with PRV gEdeficient vaccine strain and positive sera for antibodies against PRRSV,PCV2,TGEV,and sHEV.
The reproducibility of bELISA was assessed by testing three positive and three negative pig sera.These six samples were used to perform the intra-assay and interassay variabilities.The coefficient of variation (CV) was used to evaluate the inter-assay variation (between plates) and the intra-assay variation (within a plate).Each sample was tested using three different plates tested on different occasions to determine the inter-assay CV,and three replicates within each plate were used to calculate the intra-assay CV.
To evaluate the stability of bELISA following the production of the commercial kit,the coated plates and nanobody-HRP fusion protein were stored at 4°C and analyzed.The plates were coated with the purified recombinant PRV-gE (2 μg mL-1,100 μL per well) for 1.5 h at 37°C and then blocked with the blocking buffer.After that,the plates were dried and stored at 4°C under a vacuum.Meanwhile,the fusion proteins were also stored at 4°C.Then,the stored plates and fusion proteins were applied to perform the direct ELISA and bELISA every 15 d.
2.10.Comparisons between the blocking ELlSA and the commercial ELlSA Kit for testing the pig sera
To determine the coincidence of the bELISA with the commercial ELISA Kit,233 clinical pig sera were tested using two assays.Then,the inconsistent sera between bELISA and commercial ELISA Kits were further tested using IFA which was universally used to identify the PRV isolation as the gold method (Romeroet al.1997;Zhanget al.2019;Xuet al.2023).Therefore,the IFA using the PRV-infected PK-15 as the antigen and pig sera as the primary antibodies were performed to confirm the pig sera contanining anti-PRV antibodies.Briefly,the PRV (strain Ea)-infected PK-15 cells (1 MOI) were fixed with 4% paraformaldehyde (Sigma Aldrich,USA),permeated with 0.25% Triton X-100 (Sigma Aldrich),and then blocked with 1% BSA.The inconsistent serum samples diluted with 1:20 were used as the primary antibodies and incubated with the fixed cells for 1 h at 37°C.Then,the FITC conjugated goat anti-pig IgG (1:500;Jackson,USA) was used as the secondary antibody.The nuclei were stained with 4´,6´-diamidino-2-phenylindole (DAPI).Finally,the stained cells were observed under a fluorescence microscope (Leica AF6000,Wetzlar,Germany).Mock-infected PK-15 cells were used as controls to assess background staining.The coincidence rates of tested results were calculated using Microsoft Excel’s CORREL function.
2.11.ldentification of the epitope recognized by the nanobody-HRP fusion protein
To define the key amino acids involved in the interaction between the nanobody-HRP fusion protein and PRV-gE protein precisely,the 3D structures of homology modeling for PRV-gE and the nanobody were generated by submitting the amino acid sequences of the two proteins to the AlphaFold2 server (Jumperet al.2021).The docking model of the interactions of the two proteins was then developed using the docking program on the server ClusPro (cluspro.bu.edu/home.php) (Kozakovet al.2017).Interaction sites were analyzed using PyMOL (pymol.org/2/support.html) (Kagamiet al.2020).
Subsequently,the predicted key amino acids were separately mutated to alanine (A) residues for PRV-gE to identify the predicted results.The mutant was amplified by overlap PCR with primer pairs pET28a-gE-F-BamHI (5´-CGCGGATCCATGGAGGCCGGCGACGATGA-3´),pET28a-gE-245-320-R (5´-GGCGGCAGGTAGTCGCC GATGCCCATCGCCGGGGCCGCGGGGACGCAGGCCG CGCCCAGCACCAGGTCCGGGTGGC-3´) and pET28a-gE-298-320-F (5´-GGCATCGGCGACTACCTG CCGCC-3´),pET28a-gE-498-618-R-HindIII (5´-CCCAA GCTTCGAGTCGCCCATGTCCGAGACCACGCGCGGC ATCAGGTCGAACGTGTCCCCGGGCGAGAAGAGCG CCGACGCGAACGCGAGCGCGTGGAACGCGGGCGC GTGGCGCGCGGGGCCCACCGGG-3´).The mutated PRV-gE protein was expressed as the coating antigen and the nanobody-HRP fusion protein as the testing antibody was performed to confirm the predicted results.
Furthermore,the amino acid sequences of the antigenic domains from different strains in GenBank were aligned to analyze the conservation of the epitope using the Clustal W module of Lasergene 7.1 (DNAStar,MegAlign).The different PRV strains were SC,LA,Ea,Fa (clade 2.1),HNX,HNB (clade 2.2),and ADV32751 (clade 1)(GenBank accession nos.KT809429,KU552118,KU315430,KM189913,KM189912,KT824771,and KU198433,respectively).
2.12.Statistical analysis
Kappa index values were calculated to evaluate the coincidence between bELISA and the commercial ELISA Kit using SPSS software (version 20,http://www.spss.com.cn).
3.Result
3.1.Expression and purification of the recombinant PRV-gE
SDS-PAGE analysis showed that the recombinant PRVgE with His-tag was successfully expressed as a soluble form with an expected size of 28 kDa (Fig.2-A).After the proteins were purified using a Ni-NTA resin column,the results showed that the highest purity of recombinant PRV-gE was obtained (Fig.2-B).In addition,Western blotting analysis revealed that the recombinant PRVgE reacted specifically with a positive pig serum sample for anti-PRV antibodies (Fig.2-C).So the purified recombinant PRV-gE was used to immunize the camel and as the coated antigens for screening the nanobodies and developing the bELISA.
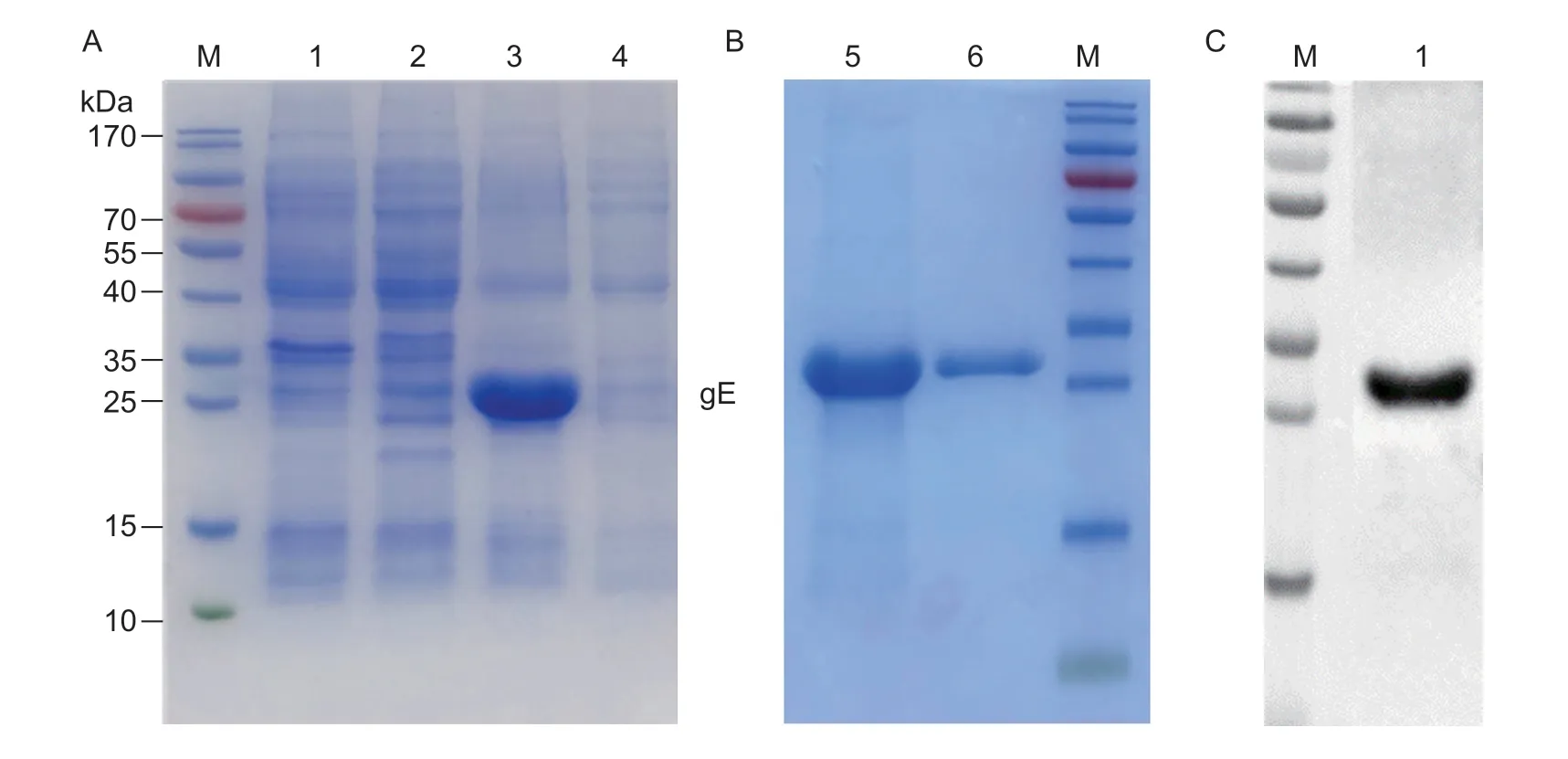
Fig.2 Expression and purification of the recombinant Pseudorabies virus glycoprotein E (PRV-gE).A and B,SDS-PAGE analysis of the recombinant PRV-gE expressed by Escherichia coli system.M,marker;lane 1,blank pET-28a vector control;lane 2,noninduced bacterial lysates;lane 3,soluble protein in the supernatant after sonication;lane 4,inclusion body in precipitate after sonication;lane 5,purified protein;lane 6,protein after dialysis.C,antigenicity analysis of the recombinant PRV-gE protein by Western blotting.M,marker;lane 1,purified protein.
3.2.Construction of the VHH library and screening of nanobodies against PRV-gE
The indirect ELISA results showed that the antibody titers against PRV-gE reached 1:107after the camel was immunized five times (Fig.3-A).Subsequently,a phage display VHH library consisting of approximately 7.85×108individual clones was successfully constructed using the PBLs from the immunized camel.In addition,48 clones were randomly picked to check the insertion rate of VHH genes by PCR,and the positive rate was 100% (Fig.3-B).
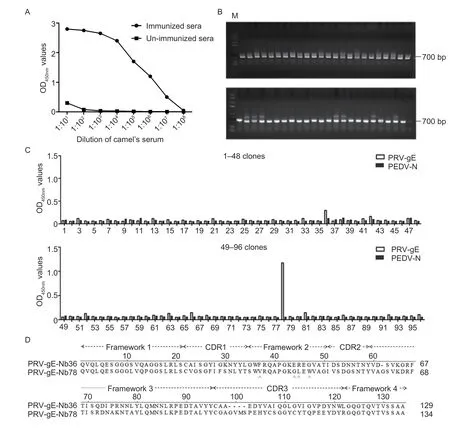
Fig.3 Construction of the VHH library and screening nanobodies against the recombinant pseudorabies virus glycoprotein E (PRV-gE).A, titers of antibodies against PRV-gE protein in the sera from the immunized camel.B,48 clones were randomly picked to estimate the correct insertion rate by PCR.C,identification of the periplasmic extracts from the 96 clones specifically binding to the PRV-gE protein by indirect ELISA.Only two clones were identified as positive.PEDV-N was used as an irrelevant protein control.D,alignment of the amino acid sequences of two nanobodies against the PRV-gE.The sequences are grouped according to their CDRs.
After three rounds of bio-screening,the phage particles against PRV-gE were significantly enriched (Table 1).The indirect ELISA results showed that 2 nanobodies specifically reacted with the PRV-gE protein but not with the PEDV-N protein (Fig.3-C).According to the amino acid sequence of the two clones,two nanobodies against PRV-gE were screened and named PRV-gE-Nb36 and -Nb78 (Fig.3-D).

Table 1 Enrichment of phage particles against the recombinant pseudorabies virus glycoprotein E (PRV-gE) during three rounds of panning1)
3.3.Expression of nanobody-HRP fusion proteins against PRV-gE in the HEK293T cells
After the positive plasmids containing the genes encoding nanobodies with HRP and His-tags were transfected into the HEK293T cells,the IFA results showed that two nanobody-HRP fusion proteins were successfully expressed in the cells (Fig.4-A).In addition,the results of direct ELISA using the medium from the transfected HEK293T cells as the primary antibody showed that the nanobody-HRP fusion proteins were secreted into the medium and still bound to the recombinant PRV-gE,but not PEDV-N (Fig.4-B).The two fusion proteins were named PRV-gE-Nb36-HRP and -Nb78-HRP.The titers of PRV-gE-Nb36-HRP and -Nb78-HRP against PRV-gE in the medium were determined to be 1:103and 1:104,respectively,and the PRRSV-Nb did not bind to PRVgE (Fig.4-C).The direct ELISA also showed that PRVgE-Nb36-HRP has a higher affinity to PRV-gE,and the OD450nmvalue can reach more than 1.5 when diluted 1:100 (Fig.4-C).Therefore,the PRV-gE-Nb36-HRP was selected as the probe for developing the bELISA.
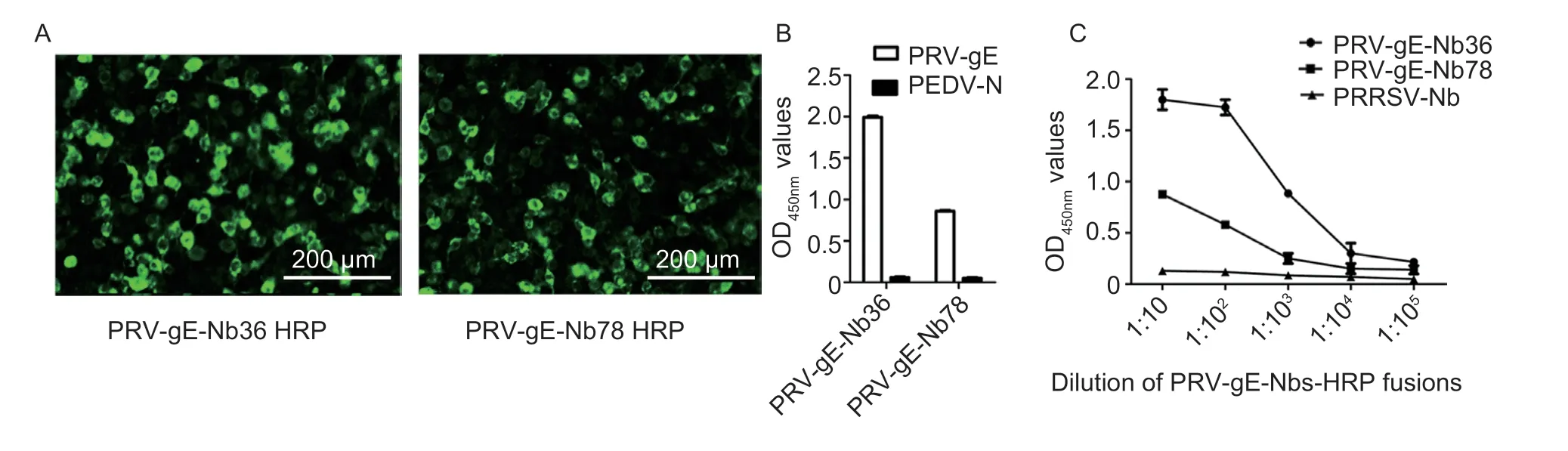
Fig.4 Expression of two PRV-gE-Nbs-HRP fusion proteins in the HEK293T cells.A,identification of two PRV-gE-Nbs-HRP fusion proteins in the cytoplasm of HEK293T cells by IFA.Anti-His monoclonal antibody was used as the primary antibody.B, analysis of specific reactions between the two screened nanobodies and PRV-gE protein by direct ELISA.C,titers of the PRV-gE-Nb36-HRP and -Nb78-HRP fusions in the medium of HEK293T cells using direct ELISA.
3.4.Establishment of blocking ELlSA for detecting anti-PRV-gE antibodies using PRV-gE-Nb36-HRP fusion protein as a probe
By the checkboard titration assay,the optimized amount of recombinant PRV-gE as the coating antigen for the bELISA was 200 ng per well,and the best dilution of PRV-gE-Nb36-HRP fusion protein was 1:320 (Table 2).The best one for diluting pig sera in the bELISA was 1:5 (Table 3).Additionally,the results of another checkerboard assay for optimizing the incubation times showed that the times of pig sera with the coated plates and of PRV-gE-Nb36-HRP fusions with the plates after incubation with pig sera were separate 50 and 30 min (Fig.5-A and B).The time of the colorimetric reaction was 10 min (Fig.5-C).
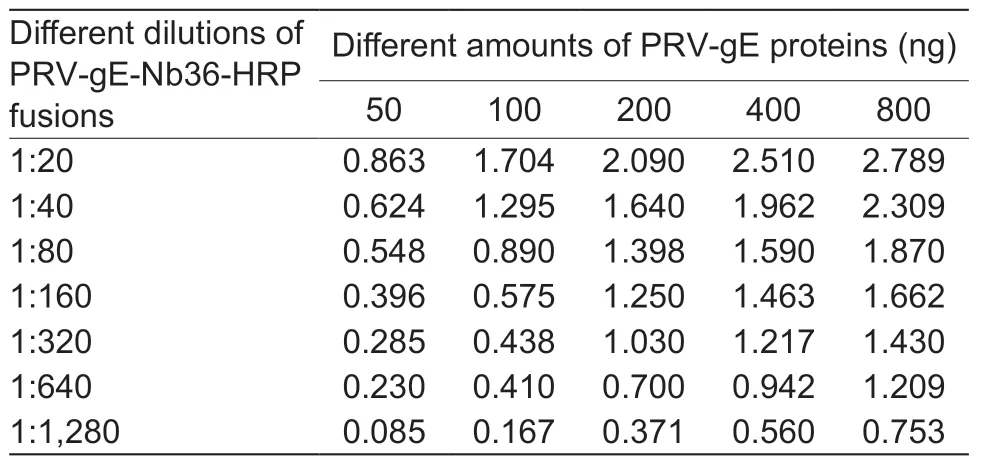
Table 2 Optimized amounts of pseudorabies virus glycoprotein E (PRV-gE) as coated antigen and dilution of nanobody-HRP fusion protein in the bELISA for detecting anti-PRV-gE antibodies
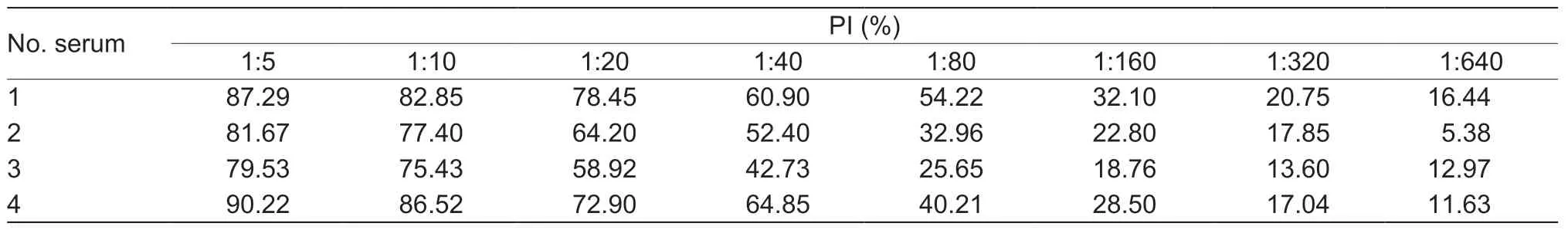
Table 3 Optimized dilution of tested pig sera for the blocking ELISA (bELISA)
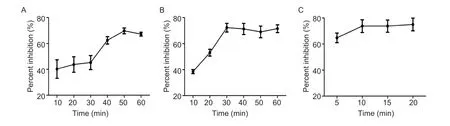
Fig.5 Optimized incubation times for the blocking ELISA (bELISA) to detect anti-PRV-gE antibodies in the pig sera.A,optimized incubation time of pig sera with the coated plate.B, optimized incubation time of PRV-gE-Nb36-HRP fusion protein with the coated and incubated plate by the pig sera.C, optimal time for the colorimetric reaction after adding TMB.
3.5.Cut-off value of the blocking ELlSA
To determine the cut-off value of the bELISA,108 negative pig sera for anti-PRV-gE antibodies were tested.The results showed that the average PI value of these negative pig sera was 9.7%,with an SD of 4.8%.So,the cut-off value of the bELISA was 24.20% (9.7%+3×4.8%),indicating that a pig serum sample with PI≥24.20% is considered positive for anti-PRV-gE antibody,and conversely,the sample is negative.
3.6.Sensitivity,specificity,reproducibility,and stability of the blocking ELlSA
To evaluate the sensitivity of bELISA,the 84 positive pig sera for anti-PRV-gE antibodies were tested.The results showed that three samples with PI values ranging from 20 to 24% were negative for anti-PRV-gE antibodies detected by bELISA.Thus,the sensitivity of the bELISA was 96.43% (Fig.6-A).In addition,the results showed that all eight positive pig sera at the dilution of 1:640 were negative by detection of bELISA,and three were negative at the dilution of 1:320 (Fig.6-B).All eight pig sera with the dilution of 1:160 were positive (Fig.6-B),indicating that the limitation of the bELISA is 1:160 for the most positive pig sera for anti-PRV-gE antibody.
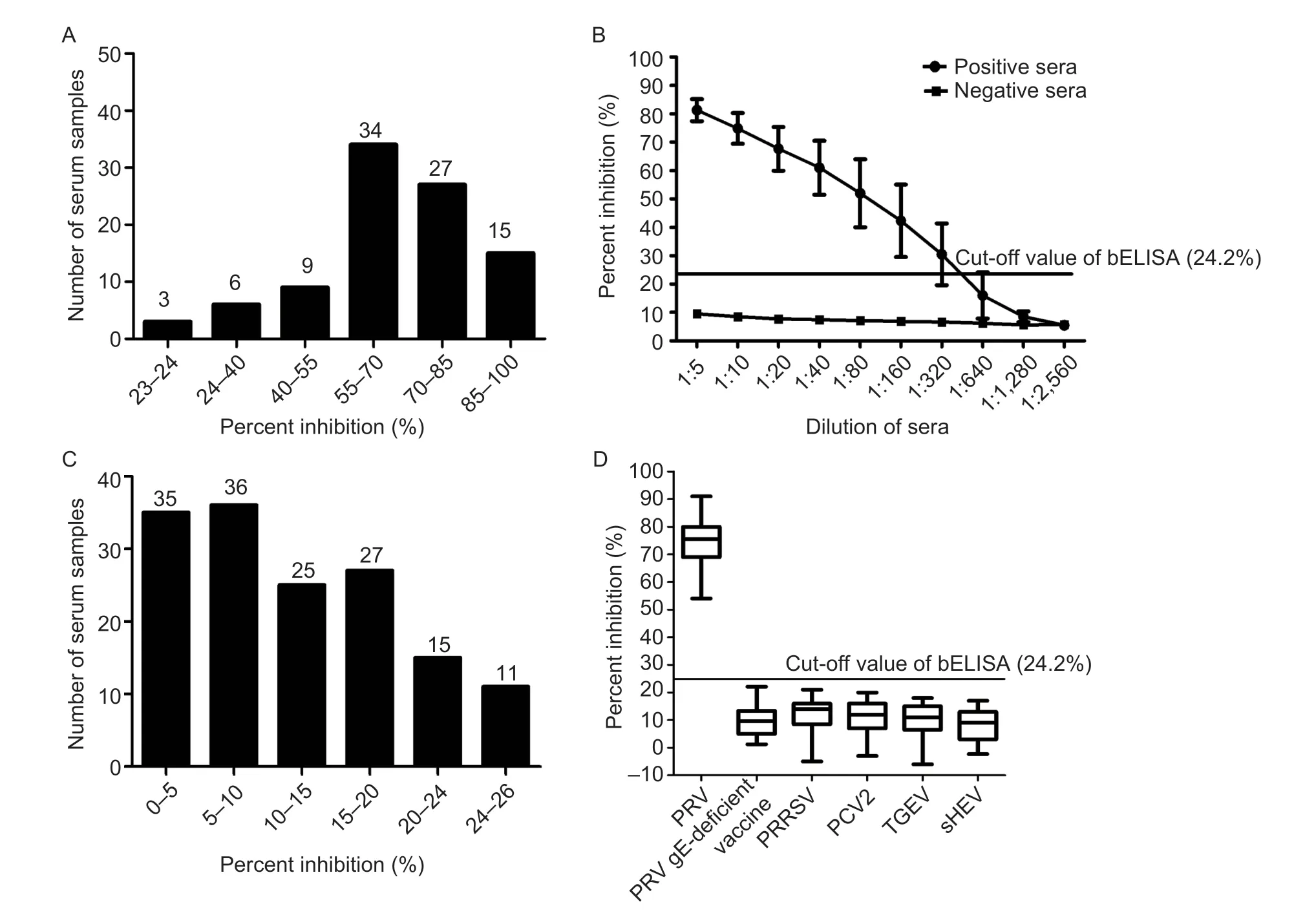
Fig.6 Sensitivity and specificity of the bELISA for detecting anti-PRV-gE antibodies.A,distribution of the percent inhibition (PI) values from the blocking ELISA (bELISA) for detecting the positive pig sera for anti-PRV-gE antibodies.B,determination of the largest dilution of positive pig sera for anti-PRV-gE antibodies detected by bELISA.C,distribution of the PI values from the bELISA to detect the negative pig sera for anti-PRV-gE antibodies.D,evaluation of the bELISA detecting pig sera from the PRV gE-deficient vaccine immunized pigs and positive for antibodies against other swine disease viruses,including porcine reproductive and respiratory syndrome virus (PRRSV),porcine circovirus type 2 (PCV2),transmissible gastroenteritis virus (TGEV),and swine hepatitis E virus (sHEV).
For the specificity of bELISA,the results showed that 138 of 149 negative pig sera for anti-PRV-gE antibodies were also negative detected by bELISA with PI values ranging 2 to 24% (Fig.6-C).Thus,the specificity of bELISA was 92.62%.Additionally,the sera from the pigs immunized with PRV gE-deficient vaccine and positive for antibodies against PRRSV,PCV2,TGEV,and sHEV were tested by the bELISA.The results showed that the PI values of all these sera were lower than the cut-off value of bELISA (Fig.6-D),suggesting that the bELISA is specific to detect anti-PRV-gE antibodies.
By testing the six pig sera in triplicate,the intra-assay CV of the PI ranged from 1.83 to 7.30%,with a median value of 4.16%.When the six samples were tested in three plates at different times,the inter-assay CV of the PI ranged from 1.39 to 6.81% with a median value of 3.39% (Table 4).These results showed that the bELISA has good reproducibility.

Table 4 Reproducibility of the blocking ELISA determined by CV% value of intra and inter assay
The binding of PRV-gE-Nb36-HRP fusion protein to the recombinant PRV-gE remained relatively stable after 100 d of unsealing the enzyme labeling plate,and the OD450nmvalue decreased slightly to around 1.0 (CV=6.01%) (Fig.7-A).Additionally,the blocking rates of three pig sera using the plates and fusion protein for the bELISA were almost unchanged in 100 d,and the CV of the blocking rates of three pig sera,respectively,were 0.80,1.37 and 1.20% (Fig.7-B),indicating that the coated plates and fusion proteins stored at 4°C have good stability for the following production of the commercial IDEXX ELISA Kit.

Fig.7 Stability of the developed blocking ELISA (bELISA) for following commercialization production.A,binding analysis of PRVgE-Nb36-HRP fusion protein to pseudorabies virus glycoprotein E (PRV-gE) at different times using direct ELISA.B,analysis of the positive pig sera for anti-PRV-gE antibodies blocking PRV-gE-Nb36-HRP fusion protein to react with PRV-gE using the bELISA at different times.
3.7.Agreement between the developed blocking ELlSA and the commercial ELlSA Kit
After a total of 233 clinical pig serum samples were detected by both the developed bELISA and a commercial IDEXX ELISA Kit,the results showed that the positive rate using the bELISA was 39.48% (92/233),and one of the commercial kits was 36.05% (84/233) (Table 5).The coincidence rate between the two methods was 93.99% (Table 5).In addition,statistical analysis showed that the results of bELISA were not significantly different from those of the commercial IEDXX ELISA Kit (Kappa=0.872).
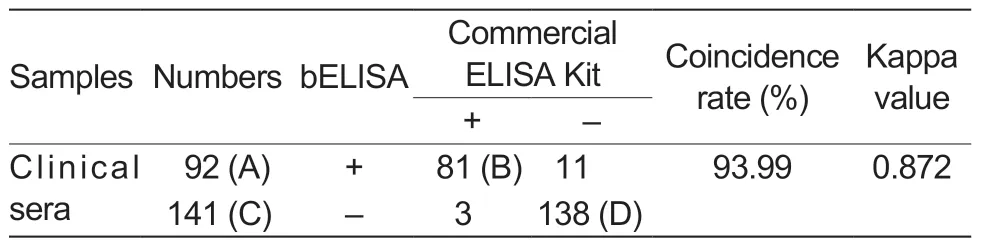
Table 5 Comparisons between the developed bELISA and a commercial ELISA Kit1)
Additionally,the 14 inconsistent sera between the two methods were further determined by IFA.The results showed that the 9 sera were consistent with the bELISA assay,and 5 were consistent with the commercial IEDXX ELISA Kit (Fig.8).
3.8.Recognition of the conserved epitope recognized by PRV-gE-Nb36-HRP among the different reference PRV strains
To precisely define the epitope,the AlphaFold2 server was used to predict the structure of proteins.The docking model showed that the amino acids R140,E145,E148,E224,R226,G231,H233 and Q235 of PRV-gE were recognized by PRV-gE-Nb36-HRP (Fig.9-A).Based on the predicted results,the amino acids were mutated into alanine (A) and expressed using theE.colisystem.SDS-PAGE and Western blotting showed that the PRVgE mutant was successfully expressed with the expected sizes (Fig.9-B and C).Furthermore,the direct ELISA results showed that PRV-gE-Nb36-HRP did not react with PRV-gE-mutant (Fig.9-D),indicating that the predicted epitope of PRV-gE recognized by PRV-gE-Nb36-HRP was correct.Furthermore,the result also suggested that the epitope recognized by the PRV-gE-Nb36-HRP was a native conformational epitope.

Fig.9 Identification of conserved epitopes recognized by PRV-gE-Nb36-HRP among the different strains.A,structure of the predicted docking complex between PRV-gE-Nb36-HRP and PRV-gE protein.The PRV-gE protein is shown in green and PRV-gENb36-HRP is shown in blue.B,SDS-PAGE analysis of PRV-gE-mutant protein expressed using the Escherichia coli system.M,protein molecular markers;lane 1,PRV-gE;lane 2,PRV-gE-mutant.C,Western blotting to identify the expression of PRV-gE-mutant protein with anti-His monoclonal antibodies.Lane 1,PRV-gE;lane 2,PRV-gE-mutant.D,determination of PRV-gE-mutant protein reaction with PRV-gE-Nb36-HRP by direct ELISA.E,sequence alignments of the key motifs binding to PRV-gE-Nb36-HRP among different PRV strains.The different PRV strains were SC,LA,Ea,Fa (clade 2.1),HNX,HNB (clade 2.2) and ADV32751 (clade 1) (GenBank accession nos.KT809429,KU552118,KU315430,KM189913,KM189912,KT824771,and KU198433,respectively).
To further analyze the amino acid conservation of the epitope,the aa R140,E145,E148,E224,R226,G231,H233,and Q235 of PRV-gE from different PRV strains were aligned.Sequence alignments showed that the epitope was highly conserved among the different PRV strains (Fig.9-E).
4.Discussion
PR remains one of the most important infectious diseases,with high mortality rates from newborn piglets and failed sow reproduction,causing huge economic losses to the pig industry worldwide.In recent years,manycountries have experimented with “marker vaccines” and launched “eradication programs” (Mettenleiter 2020).The attenuated “marker vaccines” lacking the gene encoding gE were immunized on a large scale.And then,the anti-PRV-gE antibody served as a marker to distinguish it from wild-type PRV infection.Based on this,the pigs with positive anti-PRV-gE antibodies were eliminated,thereby eradicating PR.Some traditional commercial ELISA Kits are developed for detecting anti-PRV-gE antibodies.Many countries in Europe and North America have successfully eradicated PR disease using these methods (Zhenget al.2022).However,PRV infection is still popular in some developing countries (Liuet al.2020).Because the prices of commercial ELISA Kits using polyclonal and monoclonal antibodies as reagents were high,they cannot be universally used in the field of developing countries.Thus,developing a cost-effective and simple ELISA for detecting anti-PRV-gE antibodies and eradicating PR disease in the developing countries,including China,is still imperative.The present study developed a nanobody-based bELISA to detect anti-PRVgE antibodies in the pig sera (Fig.1-C).
The assay showed high sensitivity,specificity,and agreement with the traditional commercial ELISA Kits.The bELISA established in the present study was compared with the commercial IDEXX ELISA Kit.The results showed that the coincidence rate was 96.43% for gE-positive pig sera and the coincidence rate was 92.62% for gE-negative sera.The agreement was 93.99% for detecting the clinical pig serum samples.In addition,the detection limit of the bELISA for porcine anti-PRV-gE antibody-positive serum is 1:160,which indicates that the bELISA possesses high sensitivity,especially compared to IFA which was universally used to identify the PRV isolation as the gold method (Romeroet al.1997;Zhanget al.2019;Xuet al.2023).In the study,we used the IFA to analyse whether anti-PRV antibodies existed in the pig sera.Because we found that the serum detection limit for IFA is 1:5 when the amount of inoculum is 1 MOI,the developed ELISA has good specificity and sensitivity and is promising for detecting PRV-gE antibodies in clinical pigs,creating conditions for distinguishing between vaccine-immunized and wild virus-infected pigs.More importantly,the cost of the developed ELISA was only 1/15 of the commercial ELISA Kit by calculating the cost of each reagent in the two assays.So,the nanobodybased bELISA can also be a detecting method to eradicate the PR disease by combining it with a “marker vaccine”,especially in developing countries.
Compared with polyclonal and monoclonal antibodies,nanobody has a low molecular weight,high affinity,high specificity,good stability,good tolerance,and easy productionin vitro(Wang and Wang 2022).Especially their simple genetic structure allows easy re-engineering of the protein or makes it easy to fuse with enzymes (Meiet al.2022).For example,nanobodies fused with HRP or alkaline phosphatase tags have been widely used as reagents for developing different immunoassays in diagnosing diseases and analytical methods (Zhaoet al.2022).These immunoassays can simplify the protocols and reduce the use of a secondary antibody.In the present study,we also used the PRV-gE-Nb36-HRP fusion protein as the probe to establish the bELISA.Moreover,we used DNASTAR Software to analyze the conservation of the epitope recognized by PRV-gE-Nb36-HRP among the different PRV strains.We found that the epitope was highly conserved.The developed bELISA reduces the use of secondary antibodies due to the fact that the fusion protein not only has an affinity for PRVgE binding,but also HRP has the activity of reacting with TMB,indicating that it decreases the cost of production and simplifies the operating protocols.
Additionally,chemical conjugation usually labels the polyclonal and monoclonal antibodies with enzymes.It is hard to control the molecular rations of antibodies to enzymes,which resulted in batch-to-batch variations for producing the enzyme-labeled antibodies.However,the nanobody with HRP fusion proteins was produced using the expression system with a 1:1 ratio of nanobody to HRP.This method ensured the stability of batch-to-batch in the fusion protein.Our results also showed that the titers of PRV-gE-Nb36-HRP fusion protein expressed in the different batches were the same.
Previously,the nanobodies with HRP fusion proteins have been universally preferred to develop the competitive ELISA (cELISA) because the assay was simple and had a short detection time compared with the bELISA (Shenget al.2019;Duanet al.2021;Muet al.2021).However,when we tried to establish a cELISA using the PRV-gE-Nb36-HRP fusion protein as the reagent,the results showed that the positive pig sera for anti-PRVgE antibodies and the PRV-gE-Nb36-HRP fusion protein could not compete to bind to the recombinant PRV-gE.When we developed the bELISA,the positive pig sera could block the fusion protein from binding to the PRV-gE.So,the bELISA was established using the fusion protein,although the method has a slightly longer reaction time.
Although the nanobody-based bELISA was simple compared with the traditional antibody-based bELISA,the operations of screening nanobodies were complicated.It requires the preparation of high-quality antigens,immunization of Bactrian camel,construction of phage libraries,panning for nanobodies,and expression of fusion proteins.The steps are more involved and technically demanding and may only be performed in more specialized laboratories.However,once the nanobodies have been obtained and the bELISA has been developed,the production process for the subsequent commercialization kit of the method is simple.Especially when the cell lines stably expressing nanobody-HRP fusion are constructed,it can greatly simplify the production process for the following commercial kit.Additionally,the stabilities of the coated-ELISA plate and the supernatant containing the fusion proteins were good,up to several months,with a constant blocking rate (Fig.7).Therefore,this method will have good prospects for market industrialization.
5.Conclusion
In this study,two nanobodies against the PRV-gE were screened,and nanobody-HRP fusion proteins were produced.And then,using the PRV-gE-Nb36-HRP fusion protein that recognized conserved epitopes as the probe,a novel,simple and low-cost bELISA for detecting anti-PRV-gE antibodies was developed.The bELISA showed high sensitivity,specificity,and good agreement with the commercial ELISA Kits and can distinguish between wildtype virus-infected pigs and vaccinated pigs with PRVgE-deficient strain.The assay may be an ideal method to replace the commercial ELISA Kit for detecting anti-PRV-gE antibodies and cleaning up PRV infection in pigs,especially in developing countries.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32273041),the Key R&D Program of Shaanxi Province,China (2022NY-104),and the Natural Science Foundation of Shaanxi Province,China (2022JC-12).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Ethical statement
This research did not involve animal ethics experiments.
杂志排行

Journal of Integrative Agriculture的其它文章
- OsNPF3.1,a nitrate,abscisic acid and gibberellin transporter gene,is essential for rice tillering and nitrogen utilization efficiency
- Fine mapping and cloning of the sterility gene Bra2Ms in nonheading Chinese cabbage (Brassica rapa ssp.chinensis)
- Basal defense is enhanced in a wheat cultivar resistant to Fusarium head blight
- Optimized tillage methods increase mechanically transplanted rice yield and reduce the greenhouse gas emissions
- A phenology-based vegetation index for improving ratoon rice mapping using harmonized Landsat and Sentinel-2 data
- Combined application of organic fertilizer and chemical fertilizer alleviates the kernel position effect in summer maize by promoting post-silking nitrogen uptake and dry matter accumulation