qSTA2-2,a novel QTL that contributes to seed starch synthesis in Zea mays L.
2024-05-13MinghaoCaiXuhuiLiZhiLiangJieWangDelinLiZhipengYuanRiliangGuJianhuaWangLiLi
Minghao Cai ,Xuhui Li, ,Zhi Liang ,Jie Wang ,Delin Li, ,Zhipeng Yuan,Riliang Gu,Jianhua Wang#,Li Li#
1 State Key Laboratory of Maize Bio-breeding/Key Laboratory of Crop Heterosis Utilization,Ministry of Education/Beijing Innovation Center for Crop Seed Technology/College of Agronomy and Biotechnology,China Agricultural University,Beijing 100193,China
2 Institute of Nanfan & Seed Industry,Guangdong Academy of Science,Guangzhou 510316,China
3 Institute of Crop Sciences,Chinese Academy of Agricultural Sciences,Beijing 100081,China
Abstract The seed storage materials accumulate during seed development,and are essential for seed germination and seedling establishment.Here we employed two bi-parental populations of an F2:3 population developed from a cross of improved 220 (I220,small seeds with low starch) and PH4CV (large seeds with high starch),as well as recombinant-inbred lines (RILs) of X178 (high starch) and its improved introgression line I178 (low starch),to identify the genes that control seed storage materials.We identified a total of 12 QTLs for starch,protein and oil,which explained 3.44-10.79% of the phenotypic variances.Among them,qSTA2-1 identified in F2:3 and qSTA2-2 identified in the RILs partially overlapped at an interval of 7.314-9.554 Mb,and they explained 3.44-10.21% of the starch content variation,so they were selected for further study.Fine mapping of qSTA2-2 with the backcrossed populations of I220/PH4CV in each generation narrowed it down to a 199.7 kb interval that contains 14 open reading frames (ORFs).Transcriptomic analysis of developing seeds from the near-isogenic lines (NILs) of I220/PH4CV (BC5F2) showed that only 11 ORFs were expressed in 20 days after pollination (DAP) seeds.Five of them were upregulated and six of them were downregulated in NILI220,and the differentially expressed genes (DEGs) between NILI220 and NILPH4CV were enriched in starch metabolism,hormone signal transduction and glycosaminoglycan degradation.Of the eleven NILI220 differential expressed ORFs,ORF4 (Zm00001d002260) and ORF5 (Zm00001d002261) carry 75% protein sequence similarity,both encodes an glycolate oxidase,were the possible candidates of qSTA2-2.Further analysis and validation indicated that mutation of the qSTA2-2 locus resulted in the dysfunction of ABA accumulation,the embryo/endosperm ratio and the starch and hormone levels.
Keywords: QTL mapping,seed starch,transcriptomic analysis,hormone
1.Introduction
In higher plants,starch is the important energy storage material,and it is mainly synthesized in chloroplasts of leaves (source) and amyloplasts of storage organs (library).Plant leaves perform photosynthesis during the daytime,and part of the CO2fixed by the Calvin cycle is used for starch synthesis in leaves.At night,part of the starch is degraded to produce energy,or transferred to various other cells and tissues that are incapable of photosynthesis,providing energy for plant metabolism and growth.Starch in leaves is synthesized and then degraded or transported continuously in the cycle of day and night,and the storage time is short and temporary.Seeds,tubers and roots are the main plant tissues and organs that are rich in starch.The sucrose produced by photosynthesis or the sucrose produced by the temporary degradation of starch in leaves is transported to the reservoir cells,and after a series of enzymatic reactions,starch can be formed as starch bodies for longterm storage (Huanget al.2021).Maize is an important cereal crop world-wide,playing a significant role in human and livestock nutrition (Blümmelet al.2013).The seed storage materials are accumulated during seed development,and are also essential for seed germination and seedling establishment.As the most abundant storage carbohydrate in maize kernels,starch provides a major source of calories for humans and animals.
Several early studies demonstrated that the negative correlation between grain starch and oil contents resulted in the reduction of starch content and grain yield along with an increase in the oil content (Woodworthet al.1952;Dudley 1974;Lambert and Hallauer 1994;Liuet al.2008;Cooket al.2012).Studies have also shown that the starch content is positively correlated with kernel mass,while oil content is negatively correlated with kernel mass and starch content (Wassomet al.2008).
In maize endosperm,sucrose is converted into glucose,and then starch is produced.Starch accounts for about 73% of the total weight of maize grains,of which about 25% is amylose and the rest is amylopectin (Whittet al.2002).Although the chemical structure of starch is not complex,the mechanisms of starch synthesis and accumulation are not well understood.Starch synthesis and accumulation related genes were mainly identified through quantitative-trait loci (QTL) analysis.
Among early studies in popcorn,Liuet al.(2008) first identified starch related QTL from two populations of F2:3and BC2F2,and identified theqCT1-2andqCT3-1loci which overlapped in the two populations.Later,Donget al.(2015) used a 258 recombinant-inbred lines (RILs) population of popcorn to detect the QTLs for starch concentration (CT),and obtained four QTLs among four different environments on chromosomes 3,4,5 and 9.Liet al.(2009) generated two F2:3populations with one highoil maize inbred GY220 and two normal inbreds 8984 and 8622,with 185 and 173 pairs of simple sequence repeats (SSRs) markers,respectively,and identified six protein and starch related QTLs that were located on chromosomes 5,8,and 10.Guoet al.(2013) used RILs of By804×B73 and identified 19 starch related loci with the conditioned oil and protein contents,including five QTLs located on chromosome 2 that explained 3.4-6.7% of the phenotypic variation.Zhanget al.(2015) used a population of 498 RILs and 151 SSRs to identify 31 starch related QTLs on chromosomes 1 (3 QTLs),2 (10 QTLs),4 (3 QTLs),5 (4 QTLs),6 (4 QTLs),7 (2 QTLs),8 (2QTLs),9 (2 QTLs) and 10 (1 QTL) in six environments.All 10 QTLs on chromosome 2 were located in Bin 2.03-2.09 with 263 maize inbred lines and a maize single nucleotide polymorphism (SNP) 50 array.Liuet al.(2016) identified QTLs related to kernel starch content through genomewide association study (GWAS) analysis,and obtained 77 candidate genes associated with starch synthesis within the 100-kb intervals of the leader SNPs.Later in 2018,they further identified QTLs related to starch granule size with the same populations (Liuet al.2018).A total of 79 and 88 candidate genes associated with starch length and width,respectively,were identified as being distributed on the QTL genomic regions.Among these candidate genes,six with high scores were predicted to be associated with maize starch granule size,and one candidate gene may affect the starch size.Karnet al.(2017) used 961 teosinte near-isogenic lines (NILs) of B73×parviglumisand identified two starch,three protein,and six oil related QTLs,which collectively explained 18,23,and 45% of the total variation,respectively.Linet al.(2019) used the IBM (intermated B73×Mo17) population of B73×Mo17 to identify eight starch related QTLs on chromosomes 1 (2),2 (1),3 (2),7 (1) and 9 (2).The major QTLQsta9.1in a 1.7-Mb interval on chromosome 9 was validated by allele frequency analysis in the extreme tails of a newly constructed segregating population.Wanget al.(2020) identified and cloned a major QTL,Ven1,which affects kernel texture by regulating the β-carotene content.Huet al.(2021) used six RILs that exhibit abundant diversity in starch content,which were a subset of the ROAM population,and performed regional association mapping using a set of 508 diverse maize inbred lines (AMP508).From that population,they characterized ZmTPS9 forqSTA4-2,which encodes a TPS involved in the trehalose metabolism.These findings provide some insights into the genetic architecture of starch content in maize kernels and provide informative clues for improving starch quantity and qualityviamolecular breeding.
In this study,a set of QTLs for seed storage materials were identified from two independent populations of I220/PH4CV (F2:3population) and X178/I178 (RILs).An overlapping starch related QTL locus ofqSTA2-2,a novel locus that explained 10.21% of the phenotypic variation of starch content,was selected for further study.With the combination of map-based cloning and transcriptomic gene regulation network analysis,we preliminarily anchored the target gene within 15 candidate open reading frames (ORFs),and we also found that the starch metabolism,hormone signal transduction and the carbohydrate degradation related pathways were affected in NILI220.Interestingly,we found that I220 and PH4CV,which showed a significant difference in kernel size,did not differ in embryo size.However,NILI220and NILPH4CVshowed a significant difference in embryo size in the absence of a difference in kernel size,suggesting thatqSTA2-2may ultimately lead to differences in seed starch content by affecting the size and development of embryos.Further studies involving gene cloning and validation,as well as functional studies,are required for genetic improvement and high-yield breeding with the corresponding germplasms identified in natural populations.
2.Materials and methods
2.1.Plant materials
A previously generated F2:3population from I220×PH4CV (Liet al.2018),and the RILs of X178 and its ingression line I178 were used for the raw mapping of seed chemical related traits,including the oil,starch and protein contents.The backcrossed populations of I220/PH4CV (with I220 as the recurrent parent) in each generation were introduced for starch related QTL fine mapping.The NILs of I220/PH4CV segregated with NILI220and NILPH4CVwere applied for RNA sequencing (RNA-Seq) and morphological analysis to explore the potential function of the QTL locus.Plant materials were cultivated in Hebei Province,China (39.48´N,115.97´E) during summer and in Sanya,Hainan Province,China (18.24´N,109.50´E) during winter.
2.2.Phenotypic analysis of seed morphology
Three replicates of 30 randomly selected seeds were scanned using an EPSON J221A scanner (Seiko Epson Corporation,Nagano-ken,Japan),followed by an image analysis to score the data for kernel length (KL),kernel width (KW) and kernel thickness (KT) by using the seeds identification and photoshop software package (Tuet al.2022).
2.3.Chemical analysis
The starch,protein and oil contents of the mapping populations were detected with a Fourier near infrared spectrometer and the established model for maize chemical analysis (TENSOR II,BRUKER,Germany).Meanwhile,the total seed starch of the parental lines and NILs was measured for 30 individual seeds by drying the seeds in a freeze dryer (Labconco FreeZone,Kansas City,MO,USA) and grinding them into a powder,which was then used to perform the starch measurement according to the user manual of the Total Starch Assay Kit (Megazyme,Wicklow,Ireland) (Mcclearyet al.1994).
2.4.QTL identification and epistatic interaction analysis
The collected phenotypic data were analyzed using IBM SPSS 20.0 (IBM Corp.,Armonk,NY) and the R statistical package (Nullet al.2011).The mean values of three replicates were used for the correlation analysis and QTL mapping.The coefficients of variation (CV,%) were calculated as follows: CV=s/x,wheresis the standard deviation andxis the mean for each trait.Linear regression analysis was used to determine the phenotypic correlation coefficients (Pearson’s) at the significance level ofP=0.05.
A genetic linkage map was constructed for this F2:3population and the RILs with SNP markers generated from a 6K array chip,with a total length of 1,689.8 cM and an average interval length of 1.3 cM (Liet al.2018).QTL mapping was conducted with the software QTL IciMapping 4.1 by using the inclusive composite interval mapping (ICIM) algorithm (Liet al.2007).The cut-off threshold likelihood of odds (LOD) value atP=0.05 was determined separately for each trait using the output from 1,000 permutations of the original trait data (Churchill and Doerge 1994).
The possibility of epistatic interactions between each pair of SNP markers was calculated using a two-way ANOVA test (Liet al.2007).A permutation test was conducted by randomly sampling 1,000 pairs of markers for 1,000 times to determine the genome-wide significantP-value threshold.After determining the minimumP-value observed for each set of permutations,the genome-wide threshold was set as the 5th percentile of all observed minimum values.The interactions withP-values less than the threshold value for each trait were taken as the epistatic interactions,and the adjacent SNP markers on both sides of the interaction were merged into a chromosome region (locus).
2.5.Fine mapping of the starch related QTLs
The F2:3population of I220/PH4CV was used for raw mapping,and the recombinants identified in the F2:3were subjected to backcrossing with I220 followed by selfcrossing to generate enough recombinants for markerbased selection and fine mapping.The recombinants were again backcrossed to I220 and also self-crossed for further mapping until a small interval was obtained.Finally,the locus was narrowed within 199.7 kb in the BC3F2:3,BC4F2:3and BC5F2:3populations.
2.6.Staining and color quantification
Dry mature seeds were cut along the embryo,then stained with nitrobluetetrazolium (NBT) to observe the superoxide as described by Xionget al.(2022).The same samples were also stained with 3,3-diaminobenzidine (DAB) to measure the hydrogen peroxide content,and tetrazolium (TTC) to determine the catalase (CAT) activity.Online software Image J (https://imagej.nih.gov/ij/) was used for determining the stained areas and color quantification.
2.7.RNA sequencing and data analysis
Developing seeds at 20 days after pollination (DAP) of the NILI220and NILPH4CVsegregated from BC5F2were collected for RNA extraction.Total RNA was extracted with an RNA extraction kit (Mei5bio,Beijing,China),followed by quality control (QC) with an Agilent2100.RNA-seq libraries were constructed and subjected to paired-end sequencing on the Illumina NovaSeq 6000 platform.After removing the adaptors and trimming the reads for quality control,the resulting clean data were mapped to the B73 reference genome (B73_RefGen_v4) with the program HISAT2 (Kimet al.2019),and then SAMtools was used for mapping analysis.Feature-Counts was used to count the reads mapped in the annotated gene model (Liaoet al.2014).The expected number of fragments per kilobase of transcript sequence per million base pairs sequenced (FPKM) was used to estimate gene expression levels.Genes were considered expressed at an FPKM value>0.1 as calculated by high throughout sequencing count in HTSeq software.Differentially expressed genes (DEGs) were determined using DESeq2 (https://bioconductor.org/packages/release/bioc/html/DESeq2.html) in the R language.The DEGs identified in developing seeds and seedlings were based on the criteria of a significant difference (adjustedP-value<0.05) with an absolute fold-change (FC)≥0.5.Combined Gene Ontology (GO;http://systemsbiology.cau.edu.cn/agriGOv2/) terms and enriched Kyoto Encyclopedia of Genes and Genomes (KEGG;http://kobas.cbi.pku.edu.cn/index.php) pathways were generated using the web-based programs agriGO 2.0 and KOBAS (http://bioinfo.org/kobas),respectively (Tianet al.2017;Buet al.2021).
2.8.qRT-PCR analysis
First-strand cDNAs of the above RNA samples were prepared by reverse transcription reaction using a StarScript II RT Mix with the gDNA Remover Kit (GenStar,Beijing,China).qRT-PCR was performed in triplicate for each sample using a SYBR Green I Kit (GenStar) and a QuantStudio 6 Flex system (ABI,USA).GAPDHwas used as an internal control with the primers listed in Appendix A.
3.Results
3.1.Seed chemical components in parental lines and mapping populations
Starch,oil and protein are the main storage materials in maize seeds,which also play important roles in seed development,yield formation,and seedling establishment.To explore the relationships between seed chemical components,seed development and seed vigor,we applied two bi-parental populations of F2:3and RILs of I220/PH4CV and I178/X178,respectively,and the four parents carry abundant diversity in terms of seed morphology and chemical components (Fig.1).
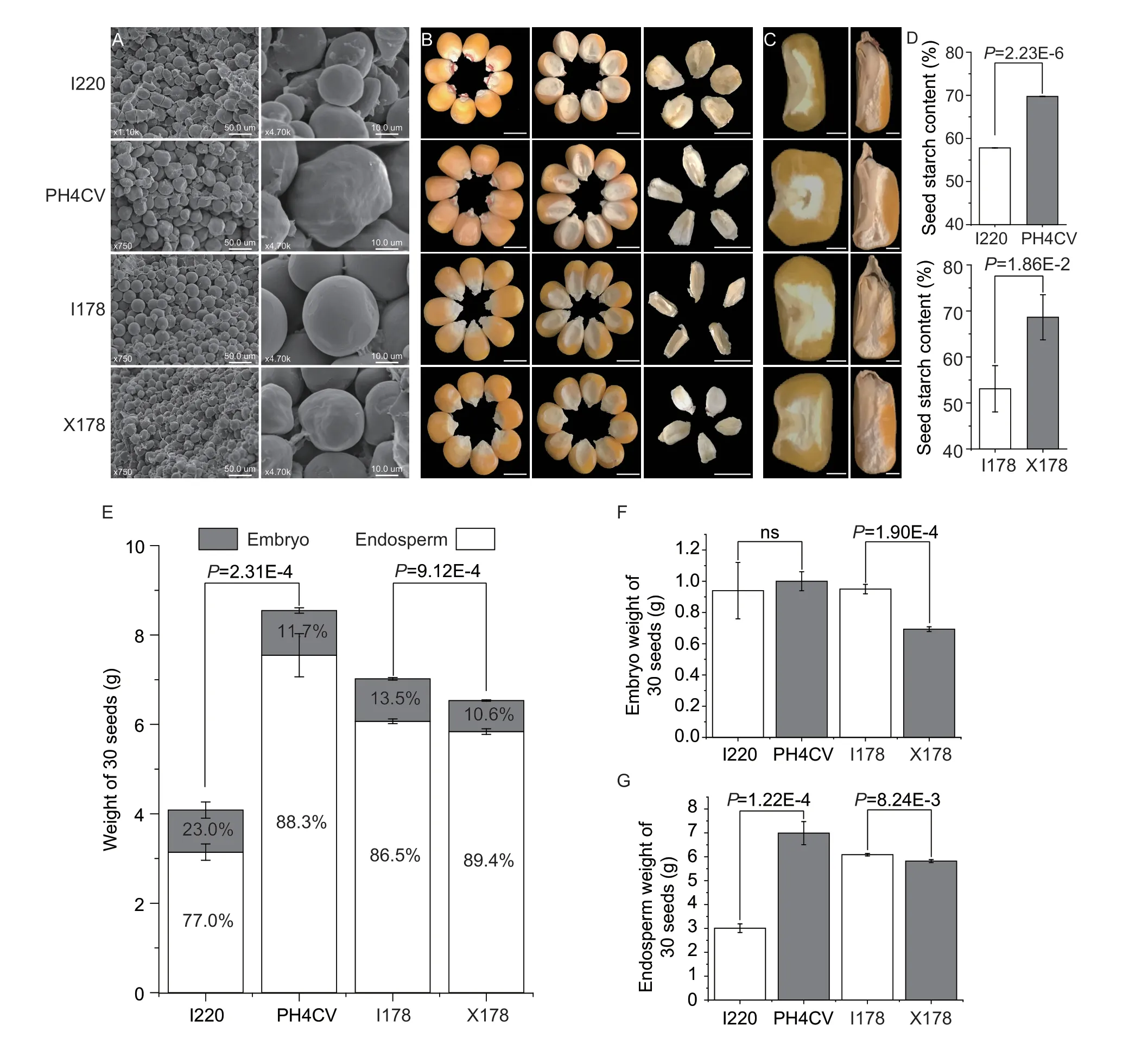
Fig.1 Physical and chemical characteristics of the parental lines.A,scanning electron microscope (SEM) observations of the starch granules in four parental lines of I220,PH4CV,I178 and X178.Bars=50.0 μm (left),10.0 μm (right).B,seed and embryo morphology.Bars=0.5 cm.C,seed cross/transections.Bars=0.1 cm.D,starch contents of I220,PH4CV (up) and I178,X178 (down) (n=10).E,30-seed weights of I220,PH4CV,I178 and X178,where the proportions of embryo and endosperm are shown as grey and white columns,respectively.F and G,embryo and endosperm weights of 30 seeds in I220,PH4CV,I178 and X178.Values in D-G represent mean±SD;no significant (ns),P>0.05;P-values were calculated by one-way ANOVA.
The oil and protein contents in I220 are significantly higher,while the starch content is significantly lower than in PH4CV.There are no significant differences in the protein and oil contents between I178 and X178,while the starch content in I178 is much lower than in X178 (Fig.1-D;Appendix B).We also analyzed the seed morphology to see whether the storage materials in seeds are related to the physical characters,and found great variation within and among the varieties.I220 has a smaller seed size than PH4CV,which was reflected by smaller values of seed length (SL),seed width (SW),seed thickness (ST),and hundred seed weight (HKW).The SW and ST in I178 are smaller,while I178 has a higher HKW than X178 due to its extremely large SL (Appendix B).Nevertheless,there is no apparent observable difference in seed size between I178 and X178,but the embryo size and shape vary considerably among the four parental lines (Fig.1-B).
With the most pronounced gradient,starch shows different patterns in the different maize lines.The starch granule sizes in I220 and X178 are smaller than those of PH4CV and I178,respectively (Fig.1-A).The proportion of embryo weight in I220 is higher than that of PH4CV,even though it has a smaller seed size,and while the embryo weight in I220 is as high as in PH4CV,the vitreous endosperm in PH4CV accounts for most of the endosperm.The proportion of embryo weight in I178 is higher than in X178,even though its HKW is lower than that of I178 (Fig.1-E-G).These findings indicated that the differences in starch contents in both comparisons were not simply explained by the seed size or weight,so more complicated mechanisms may be involved.
3.2.ldentification of QTLs related to starch,oil and protein in bi-parental populations
The F2:3populations of I220/PH4CV and RILs of I178/X178 were generated for the identification of QTLs related to starch,oil and protein.The contents of starch,protein and oil account for vast majority of seed storage materials in certain ratios,and all showed Gaussian distributions in the F2:3and RILs (Appendix C).In the F2:3populations,we identified two confident QTLs for starch (qSTA2-2andqSTA5-1),which contribute 20.42% of the phenotypic variation.Two QTLs for oil (qOIL3-1andqOIL6-1),explained 15.81% of the phenotypic variation,and three QTLs for protein (qPRO1-1,qPRO5-1,andqPRO10-1),explained 8.58-10.79% of the phenotypic variation.Five confident QTLs were detected in the RILs of I178/X178,includingqSTA2-1in Bin2.02,which partially overlapped withqSTA2-2in the F2:3of I220/PH4CV,and explained 3.44% of the phenotypic variation.Two oil related QTLs on chromosomes 9 and 10 explained 5.54-8.71% of the phenotypic variation.Two protein related QTLs on chromosomes 2 and 7 contributed 4.75-5.85% of the corresponding phenotypic variation (Fig.2;Appendix D).We also determined the epistatic effects of each QTL locus to the others,and clearly detected strong interactions between epistatic loci pairs among the chromosomes in terms of the starch,oil and protein contents (Fig.2;Appendix E).

Fig.2 QTL distribution and epistatically interacting loci for seed storage materials.A and B,QTL distribution and epistatically interacting loci for seed starch,oil and protein contents,detected from the I220×PH4CV F2:3 populations (A) and I178×X178 RILs (B).Different colored lines within the chromosome cycle represent the chromosome positions and LOD values for QTLs detected for starch,oil,and protein.Detailed QTL information is listed in Appendix D.The epistatic interactions that were found to contribute to the above three traits are labeled with corresponding colored belt curves.The width of a belt represents the width of the interacted QTL region (Appendix E).
3.3.Fine mapping of qSTA2-2 in backcrossed populations of I220/PH4CV
SinceqSTA2-2in the F2:3of I220/PH4CV was overlapping and within theqSTA2-1interval in the RILs of I178/X178,we constructed backcrossing populations of I220/PH4CV by continuous crossing of the marker based selected hybrid F1to the recurrent parent I220.In each generation,the self-crossed F2was used for genotyping and the corresponding F2:3plants were used for phenotypic analysis of the starch content to narrow down theqSTA2-2QTL interval.By using 25,000 individuals in five generations of the backcrossed populations,we finally mappedqSTA2-2into an interval of 199.7 kb (between markers DEL.37515 and DEL.37555) with nine insertiondeletion (InDel) and two SSR markers,and obtained a total of 215 recombinants,of which 111 lines had a high starch content (NILPH4CV) and 104 lines had a low starch content (NILI220).To correct our phenotypic and genotypic results,the starch contents of recombinants identified as homozygous and heterozygous in mapping were determined,and the starch contents of the heterozygous alleles varied from 55.80-70.15%,which was significantly higher than the range of the homozygous alleles (51.16-58.68%) (Fig.3-A and B).We also compared the starch contents in NILPH4CVand NILI220,and found that NILPH4CValways had a higher starch content in both developing seeds (20 DAP) and mature seeds (Fig.3-E).These results indicated that theqSTA2-2locus,which contains 14 ORFs,is responsible for the starch in this population (Fig.3-A).Comparing the seed physical characters of SL,SW,ST and HKW in NILPH4CVand NILI220,as expected,no significant differences were observed (Appendix F).After five generations of backcrossing with I220,the seed morphology of the NILs tended to be similar to I220,and the embryo proportions of NILPH4CVand NILI220were similar with no significant difference,while the embryo weight in NILI220was higher than that of NILPH4CV(Fig.3-F).These results indicated that the yield and morphology of seeds were not affected by the starch level in this population,while the embryo or endosperm proportion may contribute to the starch content (Fig.3-C and D;Appendix F).
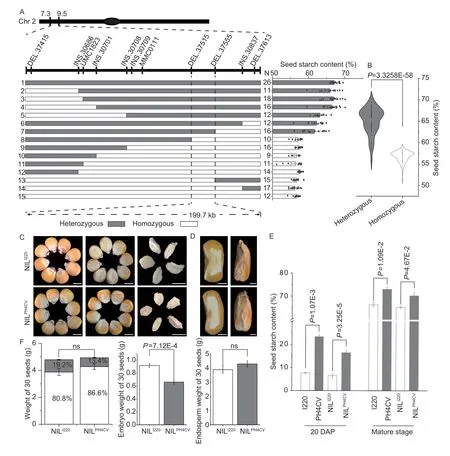
Fig.3 Fine mapping of qSTA2-2 and the performance of near-isogenic lines (NILs).A,fine mapping of qSTA2.2 with the back-and self-crossed populations of I220/PH4CV.B,violin plot of the starch contents in heterozygous and homozygous alleles used in fine mapping.C,morphological analysis of NILI220 and NILPH4CV (BC5F2:3) in terms of seeds and embryos.Bars=0.5 cm.D,seed cross/transections of NILs.Bars=0.1 cm.E,seed starch content of NILPH4CV was always higher than that of NILI220 at both 20 days after pollination (DAP) and the mature stage (n=10).F,30 seeds weights of NILI220 and NILPH4CV,where the proportions of embryo and endosperm are shown as white and grey columns,respectively.Values in B,E and F represent mean±SD;no significant (ns),P>0.05;P-values were calculated by one-way ANOVA.
3.4.Candidate gene prediction of qSTA2-2
Fine mapping identified 14 ORFs in this 199.7 kb interval (AGPv4).SinceqSTA2-2contributed to the starch accumulation in seeds,we assumed that the candidate gene should highly expressed in seeds,especially in developing seeds.We screened the tissue/organ expression patterns of those 14 genes in the qteller of Maize Genetics and Genomics database (MaizeGDB) (https://qteller.maizegdb.org/) (Woodhouseet al.2022).ORF1 (Zm00001d002257) encodes an ubiquitin-like superfamily protein,that is highly expressed in 30 DAP nonpollinated leaf,and 5 days root-cortex.ORF2 (Zm00001d002258) encodes an aminomethyltransferase1 that is also high expressed in 5 days root-cortex.ORF3 (Zm00001d002259) encodes a signal recognition particle SRP9/14 subunit that is high expressed in 12 DAP endosperm and the 5 days root elongation zone and 16-19 days vegetative meristem.ORF6 (Zm00001d002262) is also high expressed in 16 and 20 DAP embryos,V9 immature leaves,and 2-8 mm ear primordium.ORF7 (Zm00001d002266) encodes a polyamine oxidase 2 that is highly expressed in 27 DAP pericarp/aleurone,5 days root-cortex and heat treated seedlings.ORF8 (Zm00001d002267) encodes a transcription factor group E7 that is relatively highly expressed in female spikelets.ORF9 (Zm00001d002268) (disease resistance protein) and ORF10 (Zm00001d002269) (high affinity K+transporter 5) were almost not expressed in any maize tissues,with just a little bit of expression in the V18 meiotic tassel.ORF11 (Zm00001d002271) encodes an RPM1-interacting protein 4 (RIN4) that is specifically highly expressed in mature pollen.ORF12 (Zm00001d002272) (SIT4 phosphatase) is relatively highly expressed in the leaf zone.ORF13 (Zm00001d002274) (remorin family protein) is slightly expressed in ear primordium 2-8 mm.ORF14 (Zm00001d002275) (multidrug resistanceassociated protein 5) is slightly expressed in the 5 days root meristem zone.ORF4 (Zm00001d002260) encodes an aldolase-type TIM barrel family protein,is specifically expressed in seed or endosperm,and highly expressed in 12 DAP endosperm,and the paralogs ORF5 (Zm00001d002261,aldolase-type TIM barrel family protein/glycolate oxidase) that is highly expressed in 20 DAP embryo,30 DAP nonpollinated leaf,and 2-4 mm ear primordium,share 75% protein sequence similarity with ORF4,were possible the candidates ofqSTA2-2.For those 14 ORFs,only 11 ORFs were expressed in developing seeds,and six of them (ORF2,4,7,12,13 and 14) are down-regulated while the other five (ORF1,3,5,8 and 9) are up-regulated in the starch defective allele of NILI220.Furthermore,we checked the annotations of the above ORFs,combined with the above information,and further predicted that ORF4 and ORF5 are the candidates gene ofqSTA2-2(Appendix G).
3.5.Transcriptomic analysis of developing seeds in the NILs
Soluble starch synthase and starch granule-bound synthase in maize inbred lines show peaks appearing at 20-30 DAP,and the changes in amylose,amylopectin and starch accumulation rates reached their peaks at 25-30 DAP (Zhanget al.2007).To reveal the pathways and gene networks that may be affected byqSTA2-2,developing seeds of NILI220and NILPH4CVat 20 DAP were collected and RNA-Seq analysis was performed with two biological replications.We identified a total of 26,211 expressed genes in the two NILs (FPKM³0.1),among which,1,637 genes were DEGs between NILI220and NILPH4CV(|log2(FC)|≥0.5;P-value<0.05).They included 903 NILI220specifically up-regulated and 734 NILI220specifically down-regulated DEGs by comparison with NILPH4CV(Fig.4-C).GO-term enrichment analysis of all the 1,637 DEGs showed that most of the pathways were influenced in NILI220,as demonstrated by the green bars in Fig.4-A,except for the GO-terms of biological process (BP) in response to stimulus (GO:0050896),localization (GO:0051179);the GO-terms of cellular component (CC) in membrane (GO:0016020) and membrane part (GO:0044425);as well as the GO-terms of molecular function (MF) in catalytic activity (GO:0003824) and transporter activity (GO:0005215).The NILI220downregulated genes were mainly enriched in GO-terms of metabolic process (GO:0008152),biological regulation (GO:0065007),response to stimulus (GO:0050896),organelle (GO:0043226),macromolecular complex (GO:0032991),nucleic acid binding transcription factor activity (GO:0001071),transcription factor activity,protein binding (GO:0000988),nutrient reservoir activity (GO:0045735) and others (Fig.4-A).
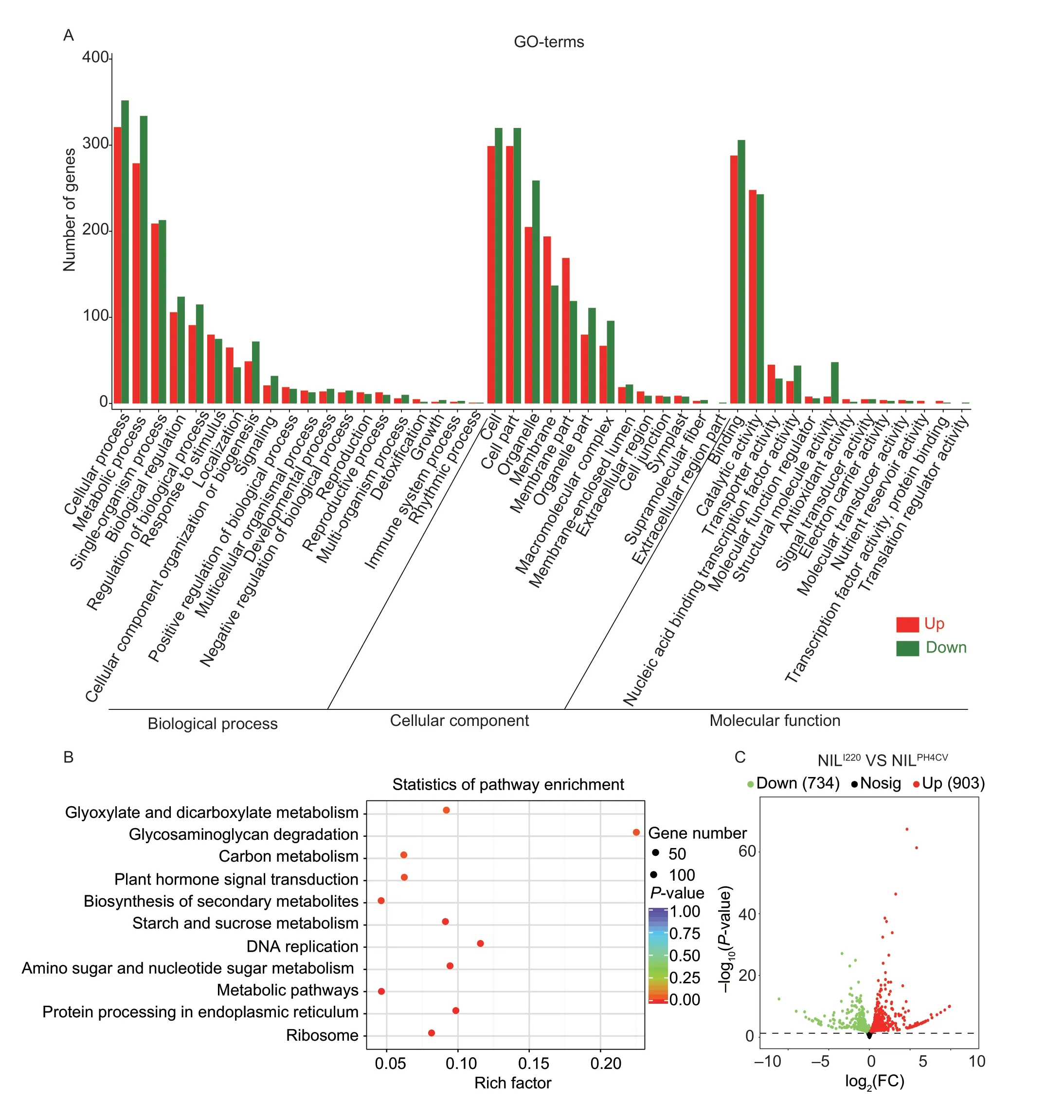
Fig.4 Gene co-expression network regulation by qSTA2-2.A,Gene Ontology (GO)-term enrichment of the differentially expressed genes (DEGs) in NILI220 and NILPH4CV detected at 20 days after pollination (DAP).The numbers of up-and down-regulated genes enriched in biological process,cellular component and molecular function are shown with red and green columns,respectively.B,Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment of the allele specific DEGs in different pathways of the near-isogenic lines (NILs).C,volcano plot showing the up-regulated (red dots) and down-regulated (green dots) genes in NILI220 and NILPH4CV.
We further focused on the top 30 most significantly enriched GO-terms based on the above information in NILI220and NILPH4CV.Of the CC-GO-terms,cytosolic ribosome (GO:00022626),perinuclear region of cytoplasm (GO:0048471),ribonucleoprotein complex (GO:1990904) and others were most enriched;for the MF-GO-terms,the ribosome structural components (GO:0003735),fructose kinase activity (GO:0008865),hexokinase activity (GO:0004396),glucokinase activity (GO:0004340) and mannose kinase activity (GO:0019158) were most enriched;similarly,the BP-GO-terms of temperature stimulation response (GO:0009266),organic matter biosynthesis process (GO:1901576),macromolecule biosynthesis process (GO:0009059) etc.,were most enriched (Appendix H).These terms indicate that the NILI220suffered somewhat from stress in the 20 DAP seeds,and the cell defense mechanisms were induced,as most components involved in biological process,cellular component and molecular function were greatly influenced.In addition,the enriched GO-terms of carbohydrate metabolism,carbohydrate kinase activity and related pathways in NILs further indicated that theqSTA2-2locus contributes to starch accumulation,with the mutation ofqSTA2-2leading to a reduction in the seed starch content in NILI220at 20 DAP.
KEGG pathway enrichment analysis revealed that 372 DEGs were significantly enriched in 11 known KEGG pathways.Among them,43 DEGs were enriched to the three most important KEGG pathways of starch and sucrose metabolism (SSM,17 DEGs),glycosaminoglycan degradation (GD,4 DEGs) and plant hormone signal transduction (PHST,22 DEGs).Besides,the processes of ribosome,protein processing in endoplasmic reticulum,metabolic pathways,amino sugar and nucleotide sugar metabolism,DNA replication,secondary metabolites biosynthesis,carbon metabolism and glyoxylate &dicarboxylate metabolism were also enriched (Fig.4-B;Appendices I and J).
3.6.Seed starch in NILs may be correlated to the hormones
After exploring the transcriptome data,we focused on the 43 DEGs enriched in starch and sucrose metabolism,as well as glycosaminoglycan degradation processes that may be regulated by the starch locusqSTA2-2.Besides,plant hormone signal transduction pathways involving the hormones of abscisic acid (ABA),indole acetic acid (IAA),cytokinin (CTK),ethylene (ETH) and jasmonic acid (JA),were also affected during seed development in NILI220(Fig.5-A;Appendices I and J).Among the 17 DEGs in starch and sucrose metabolism,six down-regulated genes encode hexokinase,β-glucosidase,and sucrose phosphate synthase that participate glucose biosynthesis/metabolism.Four DEGs were classified as associated with glycosaminoglycan degradation,of which three were down-regulated in NILI220and encode glucuronidase 3,acetyltransferase and ribosomal proteins.These results suggested that the starch synthesis and glucose metabolism activities were restrained in NILI220(Fig.5-A;Appendix I).Among the 22 DEGs involved in plant hormone signal transduction pathways,five DEGs are related to ABA signal transduction,including four downregulated genes that encode two ABA receptor-related proteins and two ABA response element binding factors;six DEGs are enriched in auxin signal transduction,including one down-regulated auxin response factor and several auxin related genes;and two downregulated genes are involved in cytokinin.All these results suggested that reduced ABA,auxin and cytokinin signaling activities in NILI220may directly or indirectly affect the starch synthesis.

Fig.5 Gene Ontology (GO)-term and Kyoto Encyclopedia of Genes and Genomes (KEGG) enriched differentially expressed genes (DEGs) in NILI220 and NILPH4CV in terms of starch,hormone and carbohydrate metabolic pathways.A,heatmap of DEGs related to starch synthesis,and hormone and sugar degradation were selected according to GO and KEGG enrichment.B,the abscisic acid (ABA) content was determined in mature seeds of parental lines and the near-isogenic lines (NILs) (n=10).C,analysis of the DEGs in starch and sucrose metabolism (SSM),plant hormone signal transduction (PHST) and glycosaminoglycan degradation (GD) by qRT-PCR and RNA-Seq (n=3).AUX,auxin;CTK,cytokinin;ETH,ethylene.Values in B and C represent mean±SD.ns,no significant;P>0.05;P-values were calculated by one-way ANOVA.
To verify whether the hormones,especially ABA,were involved in seed development or starch synthesis,we determined the ABA contents in both parental lines of I220 and PH4CV,as well as the NILs.As expected,the ABA was significantly reduced in I220 compared with PH4CV,and it was also reduced in NILI220as compared with NILPH4CV(Fig.5-B).Furthermore,the qRT-PCR analysis of the randomly selected genes in starch metabolism,hormone signal response,and carbohydrate degradation showed that these related pathways were greatly repressed,which is consistent with the RNA-Seq data (Fig.5-C).These results indicated that the regulation of storage materials in seeds is more complicated than expected,and hormones are essential for signal transduction during seed development.
3.7.Performance of seedlings and seed development in parental lines and NILs
We then wanted to determine whether the seed storage materials affected seed vigor and seedling performance.We germinated the four parental lines of I220,PH4CV,I178,and X178 and the NILs of NILI220and NILPH4CVand compared the germination speeds and rates of the above samples.At 4 days of germination,I220 showed a greater germination speed than PH4CV,as expected (Liet al.2018);while PH4CV showed better seedling performance in root length and biomass,and a final germination rate similar to I220 after 7 days,which may attributed to its larger seed size and greater amount of storage materials (Fig.6-A-E).For the NILs,there were no significant differences in germination speed or rate,in terms of either seedling roots,shoots,biomass or final rate (Fig.6-F-I).Since seedling performance is somewhat reflected by embryo vigor and reactive oxygen species (ROS) accumulation in seeds,the superoxide anion accumulation and ROS scavenging extent was stained with NBT and DAB.The results showed that PH4CV seeds accumulated more ROS and superoxide anion than I220,and similar results were observed in I178 and X178.Furthermore,the dehydrogenase activity stained with TTC,reflecting the embryo vigor,was exactly consistent with the NBT and DAB staining (Fig.6-J-L;Appendix K).In NILs,there was no significant difference in terms of ROS accumulation or embryo vigor,which was consistent with the seedling performance of the NILs (Fig.6-A,F-I,M-O;Appendix K).These results indicated that althoughqSTA2-2altered the starch content in maize,it did not influence the seed vigor or seedling performance,at least in the NILs of I220/PH4CV.
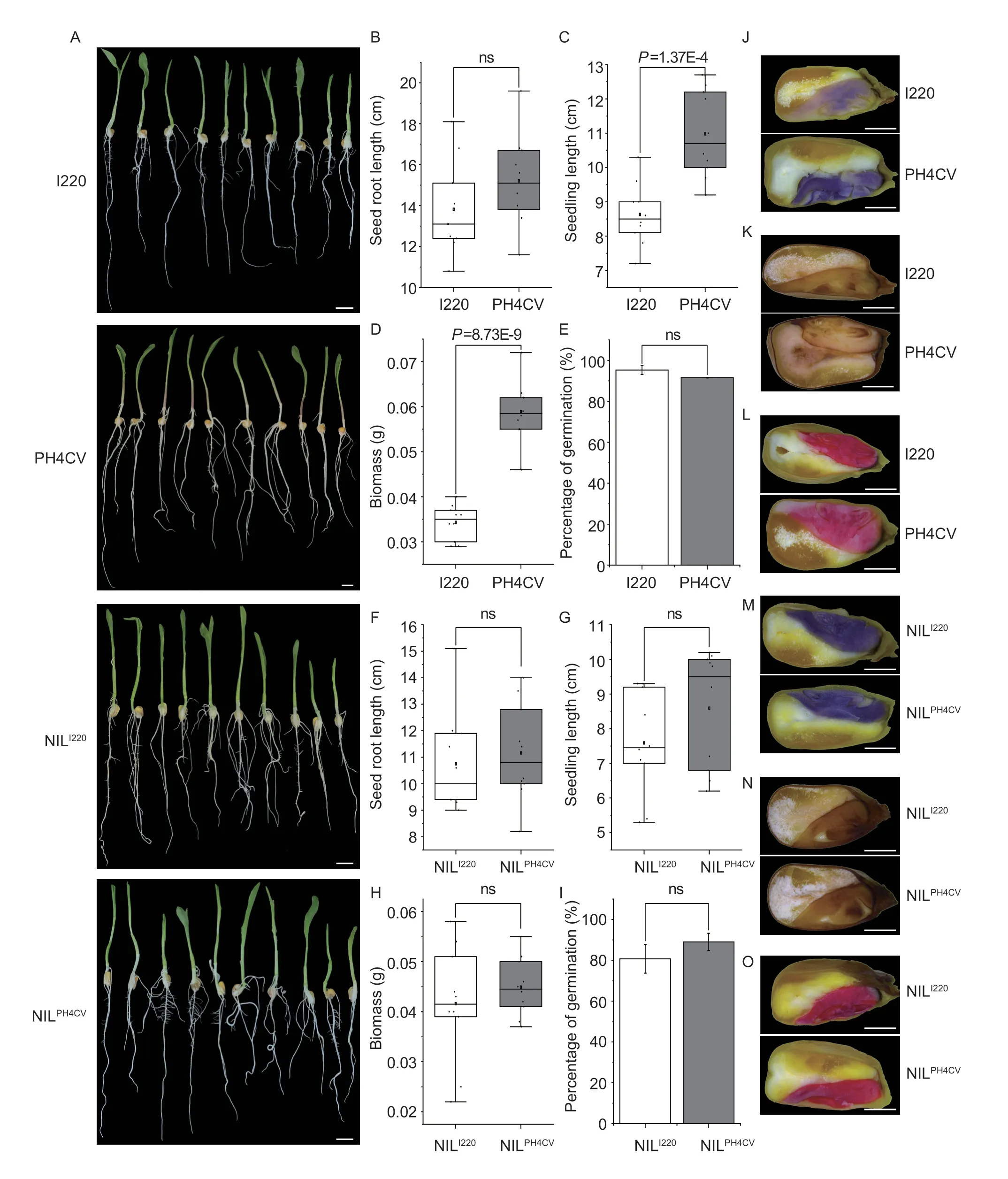
Fig.6 Physiological characteristics of seedlings and seeds in the two parental lines and NILs.A,seven-day germinated seedlings of I220,PH4CV,NILI220 and NILPH4CV.Bars=2 cm.B-I,root length (B and F),seedling length (C and G),biomass (D and H) and germination rate (E and I) of above seven-day germinated seedlings of the two parental lines (B-E) and NILs (F-I) (n=10).J-O,nitrobluetetrazolium (NBT),3,3-diaminobenzidine (DAB) and tetrazolium (TTC) staining of the superoxide anion,hydrogen peroxide and catalase (CAT) activity,respectively,in the two parental lines and NILs.Bars=0.2 cm.Values in B-I represent mean±SD.ns,no significant;P>0.05;P-values were calculated by one-way ANOVA.
4.Discussion
4.1.qSTA2-2 regulates the starch content by coordinating the embryo/endosperm ratio in seeds
Starch is the most abundant storage carbohydrate in maize kernels,and it provides calories for humans and animals.It is mainly from the endosperm of most grain seeds,a nutritional reservoir for seed germination and seedling establishment.In this study,the starch content in I220 was less than 58% and even 54% in I178 (Fig.1-D).The endosperm,which is composed mainly of starch components,accounts for 80% of the kernel mass,whereas the embryo accounts for only 10% of kernel mass,on average (Valet al.2009).Seed development is coordinated by the growth of the embryo,endosperm and maternal ovule in both monocots and dicots,in which the maternal ovule develops into the integuments and ultimately leads to the generation of the seed coat (Scottet al.1998;Garciaet al.2003;Sundaresan 2005).Interestingly,we noticed that the seed size and weight of I220 are smaller than PH4CV,but its embryo size is as large as the PH4CV embryo.The NILs were generated by continuous backcrossing with I220,so the NILs showed similar trends in terms of morphology except that the starch content was selected by markers surrounding theqSTA2-2,so it was theoretically also similar with parent I220.Although there are no significant differences in seed size or weight,the embryo of NILPH4CVis smaller than that of NILI220.This may indicate that the increasing starch content in NILPH4CVcontributed to the reduced embryo,which means an enlarged endosperm/embryo ratio.It could also indicate thatqSTA2-2may regulate both starch content and embryo size in maize.Also,I178 is an introgression line from X178,so these two have high homology in the genetic background,and the predominant seed length in I178 offset and even exceed the inferior character of seed width,so it has a higher seed weight,but still has a lower starch content than X178.In monocots and some dicots,the endosperm is retained and contributes to the volume of the mature seed.A previous study found that the growth of the seed is not primarily related to the subsequent growth of the embryo,but rather to the initial growth of the endosperm (Sundaresan 2005).Thus,I178 carries a larger embryo or smaller endosperm,which may result in the rapid proliferation of the embryonic cells in I178,or a slow proliferation of endosperm cells in X178.In summary,we concluded that three types of hypotheses may describe the seed starch accumulation in maize: Type I,large seed;type II,small embryo,and type III,small embryo but dense endosperm texture.The first two types may be attributed to theqSTA2-2effect on the embryo/endosperm ratio.More complicated mechanisms may be involved in I178 and X178 (Fig.7).One study reported that over 80% of the total kernel oil is located in the embryo,compared to only 5% in the endosperm (Lambert and Hallauer 1994).Since the oil content is negatively correlated with starch content,the ratio of embryo-toendosperm weight can partly determine the accumulation of oil.Thus,we speculated that the oil content of NILPH4CVin type II may higher than that of NILI220,although further analysis required for validation.Our results also give insight into the potential use ofqSTA2-2in crop adaptation and breeding,since it regulated the seed storage materials in NILs while not restraining the seed size or weight.
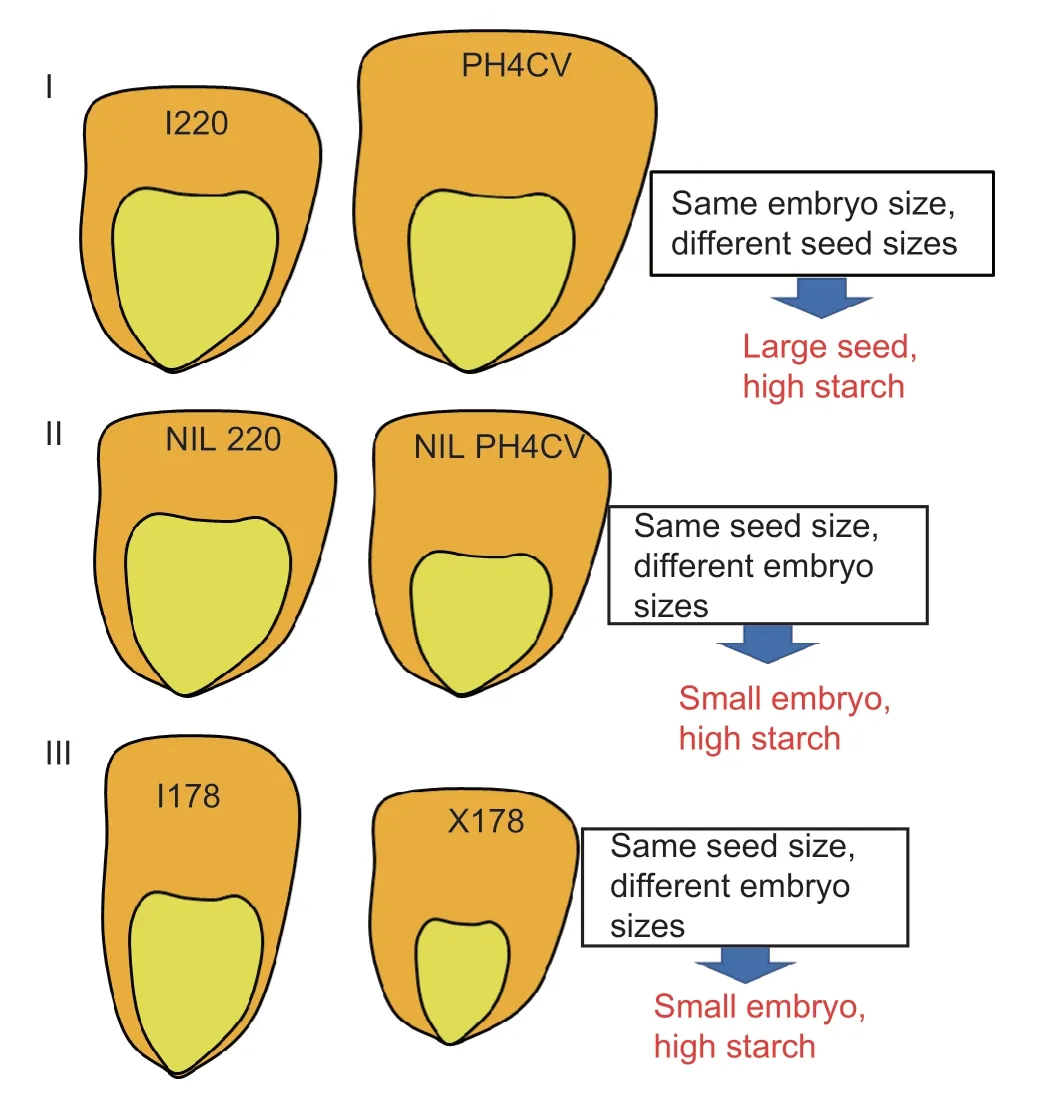
Fig.7 Three types of hypotheses that may describe the seed starch accumulation.
4.2.Starch content is not directly correlated with seed germination performance
Starch is one of the most important storage materials in seeds,since it accounts for approximately 70% of the maize seed dry weight,while protein and oil together account for less than 15% (Hannah and James 2008;Hannah and Boehlein 2017) (Appendix B).During seed germination,the endosperm of maize serves as the nutrient reservoir for seedling establishment before the three-leaf stage,and at this point the newly born leaves are able to conduct photosynthesis to provide enough energy.Thus,for a long time,the storage material,especially starch,was assumed to be correlated with seed germination ability.While this study aimed to map and clone a starch content relatedqSTA2-2,we noticed that this locus only contributed to the starch but not the seed size or weight.Furthermore,the seed germination experiment showed no significant differences between NILI220and NILPH4CV,although there were significant differences between their parental lines in terms of root length and biomass (Fig.6).To exclude the impact of seed vigor,the results of NBT,DAB and TTC staining of seed cells,which reflect the superoxide anion,hydrogen peroxide and catalase (CAT) activity,were consistent with the germination experiment.These results indicated that the variation ofqSTA2-2altered the starch content in maize seeds,while it did not affect the seed vigor or seedling establishment.However,this conclusion does not exclude the possibility of differences in seed vigor under stress conditions,since the parental lines showed apparent differences in terms of seed size and seed vigor (Liet al.2018).
4.3.Hormone signal transduction is involving in seed development and starch accumulation
Metabolites like glucose,sucrose and nitrate serve not only as nutrients but also as signals involved in the plant’s life strategies (Wobus and Weber 1999).Nakashima and Yamaguchi-Shinozaki found that abscisic acid responsive element (ABRE)-binding factor (AREB/ABF) transcription factors (TFs) regulate ABRE-dependent gene expression in seed development and germination;and the ABA signaling pathway is governed by ABA receptors and group A 2C-type protein phosphatases (PP2C) (Nakashima and Yamaguchi-Shinozaki 2013).In the GO-Term and KEGG enrichment analysis in this study,AREB/ABF2 was up-regulated while AREB/ABF1 was down-regulated in NILI220,together with the downregulation of PP2C3 and two ABI1 homologs.This was consistent with the proposition that a major ABA signaling pathway interacts with other signaling factors in stress-response and seed development (Nakashima and Yamaguchi-Shinozaki 2013).Studies have also shown that auxin levels,signaling and transport influence seed development (Figueiredo and Claudia 2018;Caoet al.2020).The plant hormone auxin has been strongly associated with numerous aspects of plant development,including seed formation (Aloniet al.2006;Overvoordeet al.2010;Locascioet al.2014;Pattisonet al.2014;Smit and Weijers 2015).GH3 genes play vital roles in auxin homeostasis by catalyzing auxin conjugation and binding free IAA to amino acids (Staswicket al.2005;Fenget al.2015;Xuet al.2020).Two of the GH3 subfamily enzymes,numbered I and II,are JA-amido,salicylic acid (SA)-amido,and IAA-amido synthetases.Previous studies have confirmed that GH3 expression is regulated by auxin,ABA,ethylene (C2H4),brassinolide (BL),gibberellins (GA),JA,SA,light and biotic/abiotic stresses (Fenget al.2015).In addition,auxin perception triggers the targeting of the AUX/IAA proteins to ubiquitination and proteasomal degradation.This degradation prevents the interaction of AUX/IAA with the auxin response factor (ARF) transcription factors.The ARFs specifically bind to auxin-responsive genes.Once free from the AUX/IAA interaction,ARFs will act either as transcriptional repressors or activators on the target genes in response to the auxin input,producing an appropriate developmental response (Luoet al.2018).In this study,five auxin-responsive GH3 family proteins were up-regulated and one auxin-responsive factor AUX/IAA was down-regulated in 20 DAP seeds of NILI220,indicating that the homeostasis of auxin ABA,ethylene,BL,GA,JA and SA may be essential for seed development,and all those hormone related pathways may participate during seed development.In addition,we enriched several ethylene signal transduction and JA related pathways,e.g.,ETHYLENE INSENSITIVE 3 (EIN3) as a negative regulator to suppress cell regeneration by inhibiting the activation of WOX genes (Liet al.2021),and found that they were also affected,including a signal transduction histidine kinase,ethylene sensor.Jasmonate ZIMdomain (JAZ) proteins act as repressors of JA signaling and regulate diverse phytohormone signalling interactions (Kazan and Manners 2012).JAZ proteins contain two highly conserved sequence regions,the Jas domain that interacts with COI1 to destabilize the repressor and the ZIM domain of unknown function,and a conserved TIFY motif (TIFF/YXG) within the ZIM domain mediates homo-and heteromeric interactions between mostArabidopsis thalianaJAZs (Chung and Howe 2009).The downregulated expression of the ethylene signal transduction and JA related genes,such as EIN3,JAZ,and TIFY domain family proteins in NILI220,indicated that the stress response system was activated or enhanced in NILPH4CVseeds.
5.Conclusion
TheqSTA2-2may regulated the seed developmentviaenergy distribution of starch,protein and other microconstituents during seed development,of which,hormones such as ABA was also involved.
Acknowledgements
This work was supported by grants from the STI 2030-Major Projects,China (2022ZD040190101,2022ZD040190502),the National Natural Science Foundation of China (32072130,32272162 and 31701437),the Project of Sanya Yazhou Bay Science and Technology City,China (SCKJ-JYRC-2023-64),the 2115 Talent Development Program of China Agricultural University,and the China Agriculture Research System (CARS-02-13).We thank Mrs.Shasha Guo for providing technical support with respect to starch analysis in X178 and I178.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2023.05.004
杂志排行

Journal of Integrative Agriculture的其它文章
- OsNPF3.1,a nitrate,abscisic acid and gibberellin transporter gene,is essential for rice tillering and nitrogen utilization efficiency
- Fine mapping and cloning of the sterility gene Bra2Ms in nonheading Chinese cabbage (Brassica rapa ssp.chinensis)
- Basal defense is enhanced in a wheat cultivar resistant to Fusarium head blight
- Optimized tillage methods increase mechanically transplanted rice yield and reduce the greenhouse gas emissions
- A phenology-based vegetation index for improving ratoon rice mapping using harmonized Landsat and Sentinel-2 data
- Combined application of organic fertilizer and chemical fertilizer alleviates the kernel position effect in summer maize by promoting post-silking nitrogen uptake and dry matter accumulation