含膦胺配体的1,2-二芳基乙烯-1,2-二硫醇镍配合物的合成、结构表征与电催化制氢性能
2024-05-07纪润武姚博馨刘梦南何佳钰赵文怀
纪润武, 何 毅, 姚博馨, 刘梦南, 何佳钰, 赵文怀, 谢 斌,,c*
(四川轻化工大学 a.材料科学与工程学院 b.化学与环境工程学院 c.材料腐蚀与防护四川省重点实验室, 四川 自贡 643000)
0 引言
氢气因其具有卓越的能量密度、高燃烧效能及其燃烧后唯一产物为水的特点,被普遍视为一种理想的、环保的、可持续的能源[1-4].采用氢气作为清洁能源代替传统碳基燃料是一项能显著减少温室气体排放的绿色环保措施.从目前来看,制备氢气的方法主要有水电解制氢、化石燃料制氢、光催化制氢[5-9]以及电催化制氢[10-13]等.其中电催化制氢是目前制氢方法的研究热点[10,14-15],它可有效降低水电解制氢的过电位,进而降低制氢的能源消耗.尽管Pt、Pd等贵金属对电催化制氢的效果十分显著,但是其价格昂贵,因此需要开发地球储量丰富的过渡金属来代替贵金属作为催化制氢的活性中心[16-17].为此,许多含钴[18]、铜[19]、镍[20-22]等金属的配合物被用作电催化制氢的催化剂.其中金属镍,不仅储量丰富,而且还存在多种氧化态形式,因此其构筑的配合物可作为电催化制氢的催化剂.研究表明,含双膦配体和二硫醇配体的过渡金属配合物均表现出良好的电催化还原质子制备氢气的性能[23-26],尤其是含氨基双膦(简称为膦胺)、1,2-二硫醇和1,3-二硫醇等配体的异配物的合成与电催化制氢性能的研究备受科学工作者的关注[27-29].因此,按照图1所示方法,本文报道了10种含膦胺配体的1,2-二芳基乙烯-1,2-二硫醇的镍配合物[(p-XC6H4N(PPh2)2)Ni(S2C2(C6H4Y-p)2)](1:X=Br,Y=CH3O;2:X=Br,Y=CH3;3:X=Br,Y=H;4:X=Br,Y=Br;5:X=Br,Y=F;6:X=F,Y=CH3O;7:X=F,Y=CH3;8:X=F,Y=H;9:X=F,Y=Br;10:X=F,Y=F)的合成、结构表征及电催化制氢性能.研究结果表明这些镍配合物都具有良好的电催化制氢性能.
1 实验部分
1.1 试剂与仪器
所有试剂均为分析纯,未作纯化处理.配体p-BrC6H4N(PPh2)2和p-FC6H4N(PPh2)2按照文献[30-32]方法改进合成,配体Ni(S2C2(C6H4Y-p)2)2按照文献[33-34]方法改进合成.
以KBr压片法在Nicolet 6700型傅里叶变换红外光谱仪上测定配合物的红外光谱.在Bruker AV 400 MHz核磁共振谱仪上测定配合物的核磁共振氢谱和磷谱.用 Smart Apex II X-射线单晶衍射仪对配合物1⋅DMF的单晶结构信息进行分析.配合物的紫外-可见吸收光谱的测试是在TU-1950紫外-可见分光光度计完成,分别用二氯甲烷或电解液作为溶剂.电化学性质是在CHI660E电化学工作站上用三电极系统(直径为3 mm的玻璃碳工作电极、铂丝对电极和非水溶液的Ag/AgNO3参比电极),以n-Bu4NPF6作支持电解质,DMF为溶剂,在氮气下进行测定,所有电位均用Fc+/Fc进行校准.所有的测试除特殊说明以外均是在室温下进行的.
1.2 配合物的合成
1.2.1 配合物1的合成
在100 mL的三口烧瓶中,加入Ni(S2C2(C6H4OCH3-p)2)2(3.318 g,5 mmol)和20 mL三氯甲烷,然后加入p-BrC6H4N(PPh2)2(5.404 g,10 mmol),在60 ℃下回流反应4 h.反应结束后加入20 mL无水乙醇,旋蒸浓缩,有绿色固体析出时停止旋蒸,抽滤收集固体.以石油醚和二氯甲烷的混合溶剂为展开剂,通过柱层析分离提纯产物,收集绿色带,蒸发溶剂得绿色固体,3.813 g,产率85%.Anal. Calcd. (%) for C46H38BrNNiO2P2S2:C,61.29;H,4.25;N,1.55;S,7.11. Found (%):C,61.28;H,4.25;N,1.55;S,7.10. IR (KBr disk,cm-1):vC=C1599 (w),1503 (s),1488 (s),1462 (m),1435 (s);vP-N-P945 (s),894 (s);vC-S740 (s).1H NMR (400 MHz,CDCl3,δ / ppm):7.90 (q,J=6.5 Hz,8H,4o-PhP),7.55 (t,J=7.4 Hz,4H,4p-PhP),7.45 (t,J=7.5 Hz,8H,4m-PhP),7.19 (d,J=8.2 Hz,4H,2o-C6H4C),7.12 (d,J=8.7 Hz,2H,m-C6H4N),6.61 (d,J=8.3 Hz,4H,2m-C6H4C),6.40 (d,J=8.5 Hz,2H,o-C6H4N),3.70 (s,6H,2CH3O). UV-Vis (CH2Cl2,λmax/ nm):317,277,231.
1.2.2 配合物2的合成
用配体Ni(S2C2(C6H4CH3-p)2)2(2.998 g,5 mmol)代替配体Ni(S2C2(C6H4OCH3-p)2)2与p-BrC6H4N(PPh2)2(5.404 g,10 mmol) 反应,得到深绿色固体3.425 g,产率81%.Anal. Calcd. (%) for C46H38BrNNiP2S2:C,63.54;H,4.41;N,1.61;S,7.37;Found (%):C,63.55;H,4.40;N,1.61;S,7.36. IR (KBr disk,cm-1):vC=C1586 (w),1508 (w),1484 (s),1435 (s);vP-N-P945(s),899 (s);vC-S746 (s).1H NMR (400 MHz,CDCl3,δ / ppm):7.90 (q,J=6.7 Hz,8H,4o-PhP),7.54 (d,J=7.2 Hz,4H,4p-PhP),7.45 (t,J=7.5 Hz,8H,4m-PhP),7.14 (dd,J=15.2,8.3 Hz,6H,2o-C6H4C,m-C6H4N),6.86 (d,J=7.8 Hz,4H,2m-C6H4C),6.40 (d,J=8.7 Hz,2H,o-C6H4N),2.19 (s,6H,2CH3). UV-Vis (CH2Cl2,λmax/ nm):315,279,231.
1.2.3 配合物3的合成
用配体Ni(S2C2Ph2)2(2.717 g,5 mmol)代替配体Ni(S2C2(C6H4OCH3-p)2)2与p-BrC6H4N(PPh2)2(5.404 g,10 mmol)反应,得到深绿色固体3.413 g,产率81%.Anal. Calcd. (%) for C44H34BrNNiP2S2:C,62.81;H,4.07;N,1.66;S,7.62;Found (%):C,62.80;H,4.07;N,1.66;S,7.62. IR (KBr disk,cm-1):vC=C1591 (w),1542 (w),1485(s),1435 (s);vP-N-P943(s),897 (s);vC-S744 (s).1H NMR (400 MHz,CDCl3,δ / ppm):7.93 (q,J=6.8 Hz,8H,4o-PhP),7.62~7.55 (m,4H,4p-PhP),7.48 (t,J=7.5 Hz,8H,4m-PhP),7.26 (s,4H,2o-C6H5C),7.15 (d,J=8.5 Hz,2H,m-C6H4N),7.06 (d,J=8.2 Hz,6H,2m-C6H5C,2p-C6H5C),6.43 (d,J=8.5 Hz,2H,o-C6H4N).31P NMR (162 MHz,CDCl3,85% H3PO4,δ / ppm):68.20. UV-Vis (CH2Cl2,λmax/ nm):311,279,231.
1.2.4 配合物4的合成
用配体Ni(S2C2(C6H4Br-p)2)2(4.295 g,5 mmol)代替配体Ni(S2C2(C6H4OCH3-p)2)2与p-BrC6H4N(PPh2)2(5.404 g,10 mmol)反应,得到深绿色固体3.016 g,产率60%.Anal. Calcd. (%) for C44H32Br3NNiP2S2:C,52.89;H,3.23;N,1.40;S,76.42;Found (%):C,52.88;H,3.23;N,1.40;S,6.41. IR (KBr disk,cm-1):vC=C1584 (w),1536 (w),1487 (s),1434 (s);vP-N-P941(s),895 (s);vC-S750 (s).1H NMR (400 MHz,CDCl3,δ / ppm):7.92 (q,8H,4o-PhP),7.60(t,4H,4p-PhP),7.50 (t,8H,4m-PhP),7.23~7.08 (m,10H,2m-C6H4C,m-C6H4N,2o-C6H4C),6.42 (d,J=8.5 Hz,2H,o-C6H4N).31P NMR (162 MHz,CDCl3,85% H3PO4,δ / ppm):67.73. UV-Vis (CH2Cl2,λmax/ nm):313,280,231.
1.2.5 配合物5的合成
用配体Ni(S2C2(C6H4F-p)2)2(3.077 g,5 mmol)代替配体Ni(S2C2(C6H4OCH3-p)2)2与p-BrC6H4N(PPh2)2(5.404 g,10 mmol)反应,得到深绿色固体3.517 g,产率80%.Anal. Calcd. (%) for C44H32BrF2NNiP2S2:C,60.23;H,3.68;N,1.60;S,7.31;Found (%):C,60.22;H,3.67;N,1.59;S,7.31. IR (KBr disk,cm-1):vC=C1545 (w),1501 (w),1485(s),1434 (s);vP-N-P943 (s),897 (s),vC-S746 (s).1H NMR (400 MHz,CDCl3,ppm):7.92 (q,J=6.7 Hz,8H,4o-PhP),7.59 (t,J=7.4 Hz,4H,4p-PhP),7.48 (t,J=7.6 Hz,8H,4m-PhP),7.24~7.12 (m,6H,2o-C6H4C,m-C6H4N),6.76 (t,J=8.7 Hz,4H,2m-C6H4C),6.41 (d,J=8.5 Hz,2H,o-C6H4N).1P NMR (162 MHz,CDCl3,85% H3PO4,δ / ppm):68.03. UV-Vis (CH2Cl2,λmax/ nm):313,278,229.
1.2.6 配合物6的合成
与1的合成方法相似,用P-FC6H4N(PPh2)2(4.795 g,10 mmol)代替P-BrC6H4N(PPh2)2与配体Ni(S2C2(C6H4OCH3-p)2)2(3.318 g,5 mmol)反应,得到深绿色固体3.423 g,产率82%.Anal. Calcd. (%) for C46H38FNNiO2P2S2:C,65.73;H,4.56;N,1.67;S,7.63;Found (%):C,65.75;H,4.56;N,1.66;S,7.63. IR (KBr disk,cm-1):vC=C1601 (w),1540 (w),1506 (s),1437 (m);vP-N-P945(s),896 (s);vC-S740 (s).1H NMR (400 MHz,CDCl3,δ / ppm):7.89 (q,J=6.4 Hz,8H,4o-PhP),7.54 (t,J=7.5 Hz,4H,4p-PhP),7.43 (t,J=7.5 Hz,8H,4m-PhP),7.20 (d,J=8.3 Hz,4H,2o-C6H4C),6.69 (t,J=8.5 Hz,2H,m-C6H4N),6.61 (d,4H,2m-C6H4C),6.42 (dd,J=8.9,4.8 Hz,2H,o-C6H4N),3.70 (s,6H,2p-CH3O). UV-Vis (CH2Cl2,λmax/ nm):317,277,232.
1.2.7 配合物7的合成
与2的合成方法相似,用p-FC6H4N(PPh2)2(4.795 g,10 mmol)代替p-BrC6H4N(PPh2)2与配体Ni(S2C2(C6H4CH3-p)2)2(2.998 g,5 mmol)反应,得到深绿色固体3.257 g,产率80%.Anal. Calcd. (%) for C46H38FNNiP2S2:C,68.33;H,4.74;N,1.73;S,7.93; Found (%):C,68.31;H,4.72;N,1.72;S,7.93. IR (KBr disk,cm-1):vC=C1541 (w),1506 (s),1476 (w),1435 (s);vP-N-P945(s),897 (s);vC-S752 (s).1H NMR (400 MHz,CDCl3,δ / ppm):7.95~7.83 (m,8H,4o-PhP),7.60~7.49 (m,4H,4p-PhP),7.43 (tt,J=7.2,1.5 Hz,8H,4m-PhP),7.17 (d,J=8.1 Hz,4H,2o-C6H4C),6.87 (d,J=7.9 Hz,4H,2m-C6H4C),6.74~6.65 (m,2H,m-C6H4N),6.42 (dd,J=8.9,4.7 Hz,2H,o-C6H4N),2.20 (s,6H,2p-CH3). UV-Vis (CH2Cl2,λmax/ nm):313,278,231.
1.2.8 配合物8的合成
与3的合成方法相似,用p-FC6H4N(PPh2)2(4.795 g,10 mmol)代替p-BrC6H4N(PPh2)2与配体Ni(S2C2Ph2)2(2.717 g,5 mmol)反应,得到深绿色固体3.422 g,产率88%.Anal. Calcd. (%) for C44H34FNNiP2S2:C,67.71;H,4.39;N,1.79;S,8.22;Found (%):C,67.70;H,4.40;N,1.78;S,8.21. IR (KBr disk,cm-1):vC=C1594 (w),1506 (s),1490 (w),1436 (s);vP-N-P947(s),900 (s);vC-S742 (s).1H NMR (400 MHz,CDCl3,δ / ppm):7.93 (q,J=8.5,6.6,1.3 Hz,8H,4o-PhP),7.61~7.54 (m,4H,4p-PhP),7.47 (tt,J=7.1,1.5 Hz,8H,4m-PhP),7.30 (d,J=1.8 Hz,4H,2o-C6H5C),7.09~7.01 (m,6H,2m-C6H5C,2p-C6H5C),6.76~6.69 (m,2H,m-C6H4N),6.45 (dd,J=8.8,4.8 Hz,2H,o-C6H4N).31P NMR (162 MHz,DMSO-d6,85% H3PO4,δ / ppm):68.07. UV-Vis (CH2Cl2,λmax/ nm):311,279,230.
1.2.9 配合物9的合成
与4的合成方法相似,用P-FC6H4N(PPh2)2(4.795 g,10 mmol)代替P-BrC6H4N(PPh2)2与配体Ni(S2C2(C6H4Br-p)2)2(4.295 g,5 mmol)反应,得到深绿色固体3.417 g,产率72%.Anal. Calcd. (%) for C44H32Br2FNNiP2S2:C,56.32;H,3.44;N,1.49;S,6.83;Found (%):C,56.31;H,3.43;N,1.48;S,6.83. IR (KBr disk,cm-1):vC=C1595 (w),1585 (w),1506 (s),1456 (s);vP-N-P949(s),900 (s);vC-S746 (s).1H NMR (400 MHz,CDCl3,δ / ppm):7.90 (q,J=6.7 Hz,8H,4o-PhP),7.59 (t,J=7.5 Hz,4H,4p-PhP),7.48 (t,J=8.0 Hz,8H,4m-PhP),7.21 (d,J=8.3 Hz,4H,2m-C6H4C),7.13 (d,J=8.3 Hz,4H,2o-C6H4C),6.73 (t,J=8.4 Hz,2H,m-C6H4N),6.48~6.41 (m,2H,o-C6H4N).31P NMR (162 MHz,CDCl3,85%H3PO4,δ / ppm):68.35. UV-Vis (CH2Cl2,λmax/ nm):311,279,232.
1.2.10 配合物10的合成
与5的合成方法相似,用p-FC6H4N(PPh2)2(4.795 g,10 mmol)代替p-BrC6H4N(PPh2)2与配体Ni(S2C2(C6H4F-p)2)2(3.077 g,5 mmol)反应,得到深绿色固体3.274 g,产率78%.Anal. Calcd. (%) for C44H32F3NNiP2S2:C,64.73;H,3.95;N,1.72;S,7.85;Found (%):C,64.72;H,3.94;N,1.71;S,7.85. IR (KBr disk,cm-1):vC=C1547 (w),1506 (s),1501 (s),1492 (sh,m),1435 (s);vP-N-P947(s),900 (s);vC-S750 (s).1H NMR (400 MHz,CDCl3,δ / ppm):7.92 (q,J=6.5 Hz,8H,4o-PhP),7.59 (t,J=7.4 Hz,4H,4p-PhP),7.48 (t,J=7.5 Hz,8H,4m-PhP),7.25~7.19 (m,4H,2o-C6H4C),6.76 (dt,J=20.3,8.7 Hz,6H,2m-C6H4C,m-C6H4N),6.45 (dd,J=8.9,4.8 Hz,2H,o-C6H4N).31P NMR (162 MHz,CDCl3,85% H3PO4,δ / ppm):68.62. UV-Vis (CH2Cl2,λmax/ nm):312,278,229.
1.3 晶体结构测定
在273 K 下,用经石墨单色化的Mo Kα(λ=0.710 73 nm)为辐射光源,把配合物1⋅DMF的晶体放入Smart Apex II X-射线衍射仪上,采用ω/2θ方法扫描,扫描范围在6.034°≤2θ≤50.016 °,共收集到41 573个衍射点,其中有7 978个为独立衍射点(Rint=0.068 5).用ShelX程序[35]解析晶体结构,使用ShelXL程序[36]来精修结构.偏离因子R1=0.050 4 (I>2σ(I)),wR2=0.105 6.在最后的差值 Fourier 图中,最高残余电子密度为0.51 e⋅Å-3,最低残余电子密度为-0.48e⋅Å-3.配合物1⋅DMF的晶体结构如图2所示,其主要的键长和键角归纳在表1(括号数字为最后一位数存在的误差)中.

表1 配合物1⋅DMF的主要键长和键角

图2 配合物1⋅DMF的晶体结构(省略了DMF和氢原子)
2 结果与讨论
2.1 配合物1⋅DMF的晶体结构描述
从图1和表1可知,配合物1⋅DMF中的1,2-二(对甲氧苯基)乙烯-1,2-二硫醇和N-对溴苯基双(二苯基膦基)胺配体与镍原子配位,形成了几乎为平面四方形的S2P2配位环境(镍原子的配位键角之和为360.6°).P-Ni和S-Ni配位键的键长均处于正常键角范围内,螯合角S2-Ni1-S1的键角(91.55(4)°)比P2-Ni1-P1的键角(74.38°)大[37].配合物中存在1个四元螯合环NP2Ni和1个五元螯合环C2S2Ni,两个螯合环的最小二次平面的夹角为9.13°.由于存在空间位阻,配体1,2-二(对甲氧苯基)乙烯-1,2-二硫醇的两个苯环与乙烯-1,2-硫醇未形成共轭π键.
2.2 谱学表征
2.2.1 红外光谱
配合物1—10的红外特征吸收峰数据如表2所示. 在894~947 cm-1出现了两个归属于膦胺配体上的P-N-P伸缩振动吸收峰[37],在740~750 cm-1的吸收峰为1,2-二芳基乙烯-1,2-二硫醇配体上C-S伸缩振动吸收峰[38].而在1 400~1 601 cm-1出现了4—5个归属苯环以及乙烯的C=C的伸缩振动吸收峰.

表2 配合物的红外特征吸收峰数据 单位:cm-1
2.2.2 核磁共振谱
配合物1—10的1HNMR测定结果表明:配合物在大约7.92 ppm的四重峰、7.57 ppm的三重峰和7.47 ppm 的三重峰应归属于膦胺配体上PPh2的苯环质子信号,而配体膦胺的p-XC6H4N片段的苯环质子信号在6.93 ppm和6.42 ppm 附近;1,2-二芳基乙烯-1,2-二硫醇的苯环质子信号在7.20 ppm和6.75 ppm附近出现,其中3和8的苯基间位氢原子和对位氢原子的信号发生重叠,而1和6的CH3O的核磁共振信号在3.70 ppm出现单峰,2和7的CH3在2.20 ppm出现单峰.
另外,还测定了配合物3—5和8—10的31PNMR,在67.73~68.62 ppm 出现了归属于膦胺配体中P原子的核磁共振单峰.
2.2.3 紫外-可见吸收光谱
图3为配合物1—10在二氯甲烷溶液中的紫外-可见吸收光谱图.配合物在310 nm、275 nm和230 nm附近出现了三组吸收带 ,其中230 nm和275 nm 附近的紫外吸收带应归属于膦胺配体和1,2-二芳基乙烯-1,2-二硫醇配体的π-π*跃迁[39],300 nm附近的吸收峰为配体内或配体间的电荷转移(ILCT/LLCT)[35-36].

图3 配合物1-10在二氯甲烷溶液中的紫外-可见吸收光谱
2.3 电化学性质
2.3.1 循环伏安
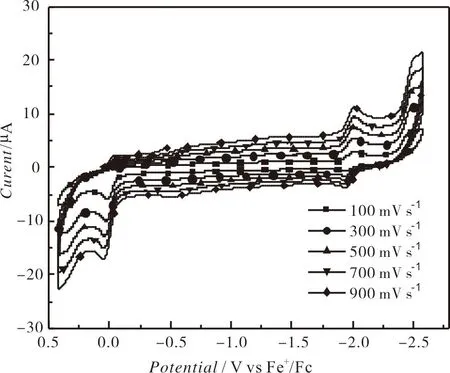
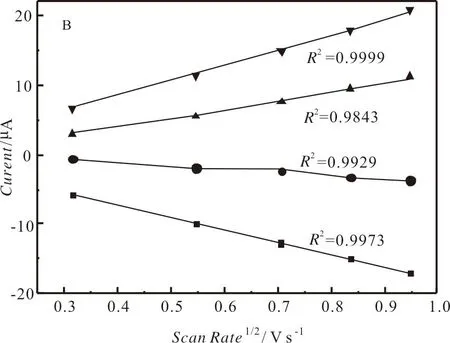
扫描速率为100 mV s-1,0.25 mM的配合物1—10在0.1 Mn-Bu4NPF6的 DMF溶液中的循环伏安图见图4,其氧化还原峰电位归纳在表3中.从图3和表3可知,配合物1—10在-1.866 V~-1.972 V(Epc1)的可逆氧化还原峰归属于NiII/NiI的单电子还原过程,而在-2.400 V附近(Epc2)的不可逆还原峰归属于NiI/Ni0的单电子还原峰[40];另外,在-0.032 V~+0.078 V的不可逆氧化峰应属于NiIII/NII的单电子氧化峰[41].分析表3中的电化学数据可知:当含有相同的1,2-二芳基乙烯-1,2-二硫醇配体时,N-(4-溴苯基)双(二苯膦基)胺配体对配合物的氧化还原峰电位正移的影响略大于N-(4-氟苯基)双(二苯膦基)胺,说明溴原子的吸电子作用略大于氟原子;当含有相同的N-(4-取代基)苯基双(二苯膦基)胺时,1,2-二(4-取代基苯基)乙烯-1,2-二硫醇配体上的取代基对配合物的氧化还原电位正移的顺序为:CH3O 表3 配合物的电化学数据 图4 配合物1-10在DMF溶液中的循环伏安曲线 配合物1在不同扫描速率下的循环伏安图如图5所示.由图5可知:随着扫描速率的提高,1的氧化还原峰的电流强度均呈现增加的趋势,其电流强度与扫描速率的平方根呈线性关系(见图6),表明1的电化学过程是受扩散控制的过程,在溶液中的活性物质可以自由扩散,相应的扩散系数(D0)为8.1×10-6cm2s-1.配合物2—10的氧化还原峰的电流强度同样与扫描速率的平方根成正比,其电化学过程也是受扩散控制的,相应的D0列在表3中. 图5 配合物1-10在不同扫描速率下的循环伏安曲线 图6 电流强度与扫描速率平方根的关系 2.3.2 电催化制氢活性 0.25 mM配合物1在扫描速率为100 mV s-1和添加不同浓度的TFA后的循环伏安图如图7所示.由图7可知:配合物1在添加5.38 mM TFA后,在-1.996 V的还原峰的电流强度增强,与NiII/NiI相比较,其还原电位向正移动了48 mV,但是其可逆性消失;而且随着TFA浓度的增加,其电流强度也随之增加,其还原电位略微发生负移,表明配合物1在TFA的存在下能催化质子还原成H2,而且气相色谱实验也证实了H2的形成.从图7可以看出,当TFA浓度为67.32 mM,催化峰的电流强度(icat)为174 μA,相应的催化峰的半波电位(Ecat/2)为-1.852 V.研究显示:配合物1加酸后的催化峰电流强度与不加酸时配合物1的还原电流比值(icat/ipc)与TFA的浓度平方根呈线性关系(见图8),表明配合物1的催化反应为一级反应.同样地,其他9个配合物也具有电催化还原制氢气的活性.配合物1-10添加67.32 mM TFA时的电化学数据总结在表3中. 图7 配合物1在不同浓度下的循环伏安曲线 图8 配合物1电流强度比值(icat/ipc)与TFA浓度平方根的关系 由表3可知,在添加67.32 mM后,配合物1-10的icat/ipc在39.77~64.60.配合物的转换频率(TOF)可以用来表征配合物催化剂的电催化活性,配合物催化剂的TOF值是通过icat/ipc的比值进行计算.由表3可知,配合物2的TOF值最高,说明配合物2的催化还原TFA制氢的效率最高.通过Fourmond方法[43],能够计算出配合物催化制氢反应时的过电位(η),从表3中可知,配合物的过电位在0.884~0.995 V.配合物4的过电位最小,这与该配合物的两个配体均含有吸电子作用最大的溴原子相一致,因此配合物4发生电催化制氢的能耗最低. 本文采用N-(4-取代基苯基)双(二苯膦基)胺与双(1,2-二芳基乙烯-1,2-二硫醇)镍配合物反应合成了10种含膦胺配体的1,2-二芳基乙烯-1,2-二硫醇镍配合物[(p-XC6H4N(PPh2)2)Ni(S2C2(C6H4Y-p)2)],并对所合成的镍配合物的结构进行了表征.测定了配合物1⋅DMF的晶体结构,其分子结构为近乎完美的四方平面结构.配合物1—10的循环伏安研究表明,配合物存在NiII/NiI的可逆氧化还原峰、NiI/Ni0的不可逆还原峰和NiIII/NII的不可逆氧化峰,这些氧化还原峰的电位值与配体所带取代基的电子效应相关联.以TFA为质子源的条件下,配合物1—10能催化TFA还原制氢,其在添加67.32 mM TFA的0.25 mM 的配合物的TOF和η的范围分别为299.32~809.67 s-1和0.867~0.995 V,其中配合物在2具有最好的催化制氢效率,配合物4的电催化制氢的能耗最低.另外,所有配合物的电化学过程都是受扩散控制的过程.因此,本文所合成的镍配合物均具有电催化制氢的能力.



3 结论
