高通量计算筛选分离乙烷/乙烯的金属有机框架材料
2024-05-06杜信明陈广慧
杜信明,陈广慧
(1.汕头大学化学化工学院,广东 汕头 515063;2.广东职业技术学院智能制造学院,广东 佛山 528500)
1 引 言
乙烯是重要的化工原材料,可用于生产聚合物和高附加值产品.截止2018 年,年产量超过1.7 亿吨[1-2],其产量可以用来衡量一个国家的石油化工发展水平.乙烯是由乙烷通过蒸汽裂解[3]方法得到的,通常伴随着5%~10%未反应的乙烷.生产高纯度的聚合物,乙烯中的乙烷杂质必须除去.乙烷和乙烯的物理性质相似,动力学直径分别为4.44 Å 和4.16 Å,沸点分别为184.55 K 和169.42 K.而C2H4比C2H6更大的四极矩,分别为1.50×10-26esu cm2和0.65×10-26esu cm2;C2H4比C2H6更小的极化率,分别为42.52×10-25cm3和44.7×10-25cm3,使得乙烷乙烯分离困难[4],被认为是世界七大分离难题之一[5].目前主要通过低温蒸馏技术获得乙烯,但是这项技术十分耗能,每年的能耗占全球的0.3%,不利于绿色工业的长远发展.因此,有必要需求固体吸附剂代替低温蒸馏技术,实现节能减排.
迄今为止,用于乙烷乙烯体系分离的固体吸附剂有碳基材料[6]、沸石[7]、多孔有机笼(POCs)[8]、金属有机框架(MOFs)[9]、共价有机框架(COFs)[10]和氢键有机框架(HOFs)[11].与活性炭、沸石等相比,金属有机框架(Metal-Organic Frameworks,MOFs)具有高孔隙率、大比表面积、可修饰位点和可调节的孔表面等独特性质,广泛应用于气体的吸附分离.目前,MOFs 用于乙烷乙烯混合物分离的策略有三种:第一种方案是基于限域效应设计合适特定孔径的MOFs,使C2H6能进入而C2H4不能进入,这无疑是十分困难的[12-13];第二种方案是选择性吸附乙烯/乙烷,这种策略通常利用开放金属位点(open metal sites,OMS)与乙烯形成反馈键,其机理主要是热力学分离[14-15],给吸附乙烯之后脱附造成一定的困难. 并且因为一步脱附含混有少量乙烷,需要多步吸附-解吸才能获得纯净的乙烯[16].例如,Qian 的小组[17]在298 K、100 kPa 下CuI@UiO-66-(COOH)2由于Cu(I)离子和C2H4之间的孔径微调和络合相互作用,选择性高达80.8;第三种方案是选择性吸附乙烷/乙烯(反向吸附),这种策略可以一步获得纯净的乙烯,不需要进行繁琐的多步吸附-解吸[16].通过MOFs 与乙烷形成比乙烯较多的氢键,或者是引入含低极性芳香环的有机配体构建非极性孔表面的MOFs,中等的乙烷吸附热有利于MOFs 的再生循环利用. 例如,Ye 等[18]研究了两种同构金属MOFs(Ni-MOF 1 和Ni-MOF 2),最终发现Ni-MOF 2 在C2H6/C2H4分离方面表现出更高的性能.通过密度泛函理论(DFT)研究表明,Ni-MOF 2 中存在未堵塞的独特芳香族孔表面与C2H6诱导出更多更强的C-H…键,而且合适的孔隙空间增强了其C2H6吸收能力;Zeng 等[19]合成的JNU-2 可以通过与乙烷形成多个C-H…O 氢键相互作用,实现从等摩尔C2H6/C2H4混合物中反向捕获C2H6,从而获得更高的C2H4生产率,约21.2 L kg-1,纯度超过99.99%. 目前,报道的大多数MOFs 是选择性吸附乙烯,而反向吸附,即选择性吸附乙烷的MOFs 还不多.因此有必要发现更多的MOFs 选择性吸附乙烷,实现一步纯化乙烯.
在本工作中,为了在298 K 和100 kPa 下获得高性能选择性吸附分离C2H6/C2H4混合物(10/90 v/v)的MOFs,根据Jiang 等[20]构建的结构-吸附性能关系(SAPR)进行了高通量筛选,工作流程如图1 所示.(1)从G-MOFs 和hMOFs 数据库中去除含有开放金属位点的MOFs;(2)使用Jiang 等[20]构建的构效关系在不含OMS 的G-MOFs[21]和hMOFs[22]数据库中进行筛选潜在高性能的MOFs,并通过GCMC 模拟验证;(3)对筛选到的高性能MOFs 的膜选择性和吸附机理进行分析和讨论.

图1 筛选吸附分离C2H6/C2H4 高性能MOFs 的工作流程
2 计算细节
2.1 蒙特卡洛模拟
在本工作中,采用GCMC 方法在μVT 系综模拟了MOFs 对C2H6、C2H4的吸附热和吸附等温线.采用通用力场(UFF)[23]描述MOFs 原子,而用TraPPE 力场[24]描述吸附质分子.Jiang 等[20]也曾使用相同的力场模拟了C2H6/C2H4分离的吸附量和选择性.在这项工作中,假定MOFs 是刚性的.对于每个GCMC 模拟,平衡和生产步数均为1×105,截断半径设为12.0 Å[25-26].在这些过程中,平移、旋转、插入和客体分子交换的比例都设置为1.0.此外,采用涨落理论(fluctuation method)[27]计算吸附热(Qst),如式(1)所示:
其中U、N、R、T 分别表示能量、粒子数、摩尔气体常数、温度.高通量计算验证是在298 K 和100 kPa的条件下,模拟C2H6和C2H4的二元混合物(10∶90)分离.而C2H6和C2H4的单组分吸附等温线是对高性能MOFs,在298 K、1.0 kPa~100 kPa 计算的.上述计算均采用RASPA 软件[28]进行模拟的.此外,根据式(2)计算C2H6/C2H4吸附选择性(SC2H4H6/C2):
其中xC2H6和xC2H4分别表示C2H6和C2H4的吸附量,而yC2H6和yC2H4分别表示C2H6和C2H4的摩尔分数,比例为10∶90.
C2H6和C2H4的亨利常数是采用Widom 插入法计算的,与上述GCMC 模拟使用相同的截断半径和力场.Widom 插入法计算采用1×105步进行计算[29].
再生能力(R%)是决定解吸过程中吸附位点的再生百分比的一个重要参数.对于混合物的分离,再生能力可通过式(3)[30]计算,是气体的工作容量与较高吸附压力下之比:
式中ΔNKr和分别代表工作容量和C2H6在100 kPa 下的吸附量.工作容量ΔNKr可以通过式(4)[31]:
最后,为了综合评估吸附剂的工作容量和吸附选择性,使用吸附剂性能打分(adsorbent performance score,APS)参数进行评价,如式(5)所示[31]:
2.2 分子动力学模拟
在298 K 下和正则系综(NVT)中,使用分子动力学(MD)模拟了C2H6或C2H4原子在MOFs 中的扩散行为.其中,总模拟时长为10 ns,步长设置为0.5 fs,前5 ns 用于平衡结构,后5 ns 为生产过程,用于计算均方根位(mean square displacements,MSD).在MD 模拟中使用了与GCMC 相同的力场参数和截断半径.此外,自扩散系数(Ds)采用爱因斯坦扩散定律[32]计算,如式(6)所示:
式中Ds和t 分别是自扩散系数和时间,是气体分子随时间t 的均方根位移(MSD).
此外,采用Materials Studio(2019)软件[33]的Forcite 模块[34]进行MD 模拟,来预测高性能MOFs 的热稳定性.使用通用力场(UFF)[23],模拟退火过程温度设置在300 K 到1 000 K之间,整个过程包括5 个循环.在NVT 系综中,总模拟时长为15 ps,步长设置为0.5 fs.
2.3 量子化学计算
首先,在密度泛函理论(DFT)计算中,采用广义梯度近似(Generalized gradient approximation,GGA)中的Perdew Burke Ernzerhof(PBE)泛函[35],双数值轨道+氢原子p轨道极化函数(double numerical plus p-functions,DNP)基组[36]和使用Grimme 提出的DFT-D2 色散[37]校正来描述每个原子的范德华作用,对高性能MOFs 吸附C2H6或C2H4后的吸附构型进行结构优化和吸附能的计算.以上计算均采用Materials Studio(2019)软件[33]的DMol3模块[38]进行,吸附能(Eads)可根据式(7)进行计算:
其中EMOF+gas、EMOF和Egas分别为MOFs 与吸附气体的复合物、空MOFs 和被吸附气体的能量.
其次,作了基于Hirshfeld 分区独立梯度模型(independent gradient model based on Hirshfeld partition,IGMH)[39]计算,探究C2H6或C2H4原子与MOFs 之间的弱相互作用.其计算流程如下:首先,采用Gaussian 16 程序包[40],使用B3LYP 泛函[41],对于轻原子使用6-31G(d)基组[42],对于过渡金属原子使用SDD 赝势基组[43]和Grimme 提出的Becke-Johnson(D3BJ)[44]进行色散校正计算波函数;然后使用Multiwfn 3.8 程序[45]对上述波函数进行IGMH 计算.
3 结果与讨论
3.1 采用构效关系高通量筛选分离C2H6/C2H4 的高性能MOFs
2020 年,Jiang 等[20]通过对CoRE MOF(2019)数据库的1 747 个MOFs,在常温常压下计算模拟C2H6/C2H4二元混合物(10∶90 v/v)的吸附分离.随后通过数据统计分析建立了构效关系,即MOFs 的物性参数与C2H6吸附性能之间的关系.当MOFs 的孔隙率为0.576~0.696、LCD 为5.6~7.5 Å、PLD 为4.2~6.1 Å 和VSA 为1 487~2 600 m2/cm3时,MOFs 能够很好地克服“权衡”问题,即同时具有较大的C2H6吸附量和C2H6/C2H4选择性.随后根据其获得的构效关系在G-MOFs 和hMOFs 数据库中筛选用于C2H6/C2H4混合物分离的高性能MOFs.
由于含有开放金属位点(OMS)的MOFs 常会因为形成反馈键而选择性吸附C2H4,吸附热远大于C2H6,从而表现出选择性吸附C2H4/C2H6. 因此,本工作从G-MOFs 和hMOFs 数据库除去含有OMS 的MOFs,然后再根据构效关系进行筛选.通过open_metal_detector[13]识别G-MOFs 和hMOFs 数据库不含开放金属位点的MOFs,最终在G-MOFs和hMOFs 数据库中,分别得到108 957 和134 187 个MOFs.然后通过Zeo++程序[46]计算hMOFs 和G-MOFs 数据库不含有OMS 的MOFs 物性参数,包括全局最大孔腔直径(Global Cavity Diameter,GCD)、孔腔极限直径(Pore Limiting Diameter,PLD)、最大孔腔直径(Largest Cavity Diameter,LCD)、晶胞体积、密度(ρ)、VSA、比表面积(Sa)和孔隙率(φ). 随后,根据上述构效关系对hMOFs 和G-MOFs 数据库不含有OMS 的MOFs 进行筛选,从hMOFs 和G-MOFs 数据库分别得到2 091 和2 157 个MOFs.
采用这种基于构效关系的筛选方式,不但计算速度快,而且节省了大量的计算资源.随后,计算Jiang 等[20]从CoRE MOFs(2019)数据库中获得的16 个高性能MOFs 对C2H6的吸附量、工作容量和SC2H4H6/C2,发现与其结果一致,表明当前的力场可靠,可以进行下一步高通量筛选.最后,在298 K 和100 kPa 下,1 kPa 两个压力点下,采用GCMC 模拟C2H6/C2H4(10/90)混合物的分离,进一步验证上述得到的4 248 个MOFs 对C2H6/C2H4吸附分离的性能. 发现了6 种高性能MOFs,即Al2O6-AZO_B-fum_B_No65、Al2O6-DPAC_B-fum_B_No152、MIL53Cr-fum_B-irmof10_B_No8、hMOF-18883、hMOF-19548 和hMOF-37529.这6 个MOFs 对C2H6吸附量较大,分别为1.33、1.40、1.05、1.55、1.71、1.50 mmol/g,IAST 选择性很大,分别为2.28、2.29、2.12、2.02、2.06 和1.99.其具体物性参数、吸附量、吸附热和选择性如表1 所示.
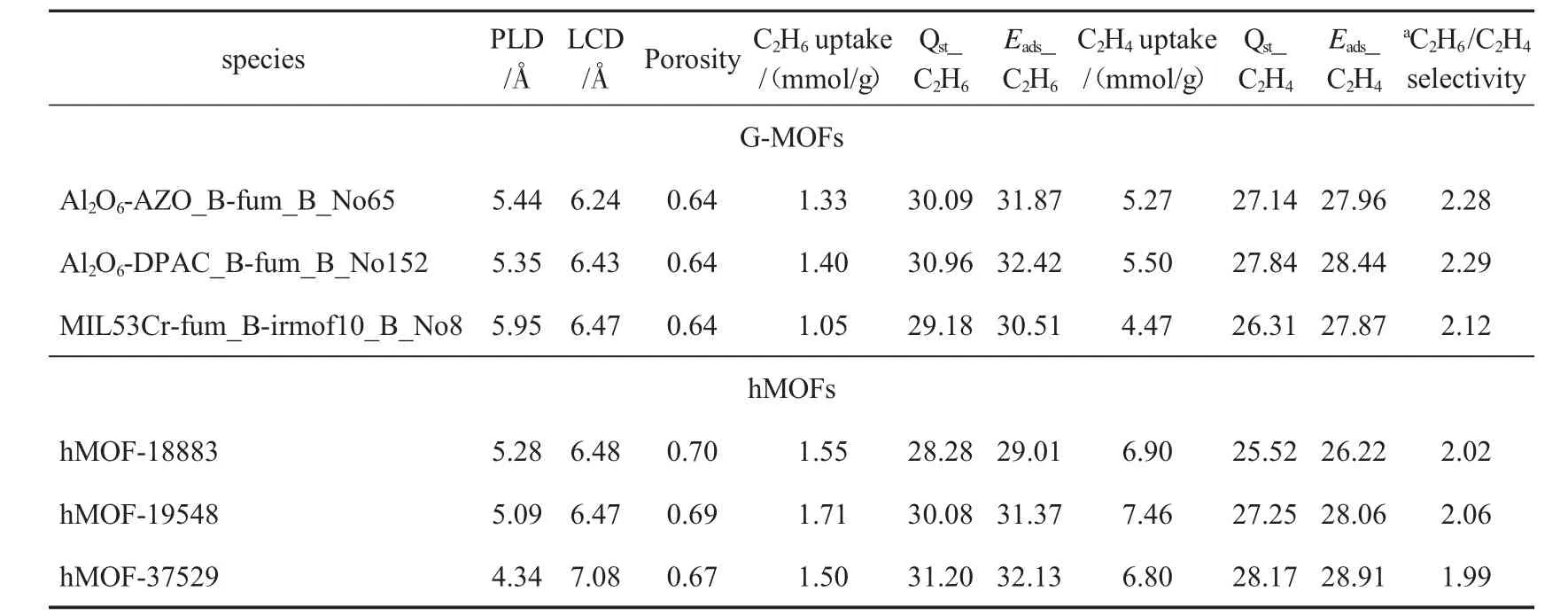
表1 在298 K 和100 kPa 下,高性能MOFs 的物性参数,在GCMC 水平C2H6∶C2H4=10∶90 条件计算的C2H6、C2H4 的吸附量、吸附热(Qst)、C2H6/C2H4 选择性和DFT/PBE 水平计算的吸附能(Eads).
3.2 分离C2H6/C2H4 高性能MOFs 的结构特点
为了深入了解选择性吸附C2H6的MOFs 的结构特点,首先观察不具备OMS 的MOFs 选择性吸附C2H6的效果,再观察吸附性能优异MOFs 的有机配体特点.
首先,对不含有OMS 的MOFs 进行GCMC 模拟,计算其IAST 选择性,如图2(a)所示.对于G-MOFs 数据库中2 157 个不含OMS 的MOFs,只有99 个MOFs 的处于0.5~1.0 之间,表明是选择性吸附C2H4;而选择性处于2.5~3.0 之间的仅有7 个,大部分MOFs 的处于1.5~2.0 之间. 如图2(b)所示,对于hMOFs 数据库2 091个不含有OMS 的MOFs,其均大于1.0,表明是选择性吸附C2H6,且大部分MOFs 处于1.5~2.0 之间.由此可见,不含OMS 的MOFs 有利于选择性吸附C2H6.

图2 GCMC 模拟SC2H6/ C2H4 的分布(a)2 157 个G-MOFs 和(b)2 091 个hMOFs.
然后,对G-MOFs 和hMOFs 数据库吸附性能排名前20 的MOFs 的有机配体进行结构统计分析,如图3 所示.发现这些有机配体不含极性基团且含的苯环数目较多.推测有利于这类有机配体的极性较低,能够构建非极性的MOFs 孔表面,从而有利于四极矩小的C2H6优先吸附.

图3 G-MOFs 和hMOFs 数据库中C2H6 吸附性能排名前十的MOFs 的连接体.
3.3 吸附机理以及电子结构分析
3.3.1 热稳定性、吸附等温线、吸附构型和膜选择性
首先,通过MD 模拟探索如表1 所示的6 种高性能MOFs 的热稳定性.在100 kPa、300 K 至1 000 K 的范围内,进行了由5 个循环组成的模拟退火,发现上述6 个MOFs结构没有塌陷,表明其结构稳定.此外,根据式(3)计算了上述6 种MOFs 在C2H6/C2H4吸附分离上的再生能力,发现其再生能力(R%)均大于50%,如表2 所示.表明它们在实验上能被较好地循环利用.6 种高性能MOFs 的晶胞结构如图4 所示,值得注意的是,目前尚未有其用于C2H6/C2H4吸附分离的报道.
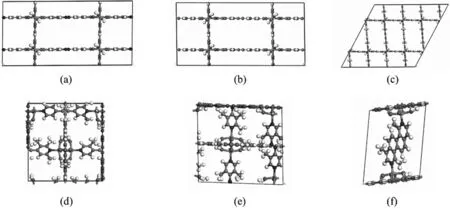
图4 用于C2H6/C2H4 吸附分离的6 种高性能MOFs 的晶体结构图,分别包括(a)Al2O6-AZO_B-fum_B_No65,(b)Al2O6-DPAC_B-fum_B_No152,(c)MIL53Cr-fum_B-irmof10_B_No8,(d)hMOF-18883,(e)hMOF-19548 和(f)hMOF-37529.

表2 在298 K 和100 kPa 条件下,计算筛选和实验报道的高性能MOFs 的C2H6 的吸附量、C2H6/C2H4选择性、工作容量(N)、再生性(R%)和APS.
其次,计算了上述6 种高性能MOFs 吸附C2H6和C2H4的亨利常数,如表3 所示.亨利常数表示在无限稀释条件下客体分子和框架之间相互作用的强度.其中,C2H6的亨利常数在29.87~133.83,而C2H4的亨利常数在13.52~52.56,亨利常数的比值为1.97~2.55.亨利常数的比值与表1 所列的IAST 选择性趋势一致,表明上述6 个高性能MOFs 与C2H6的亲和力大于C2H4,表现为选择性吸附C2H6.例如,Al2O6-DPAC_B-fum_B_No152 的C2H6/C2H4的亨利常数为2.55,IAST 选择性为2.29.亨利常数越大,客体分子和框架之间的相互作用就越强.

表3 采用Widom 插入方法计算的高性能MOFs 对C2H6 和C2H4 的Henry 系数.
然后,在298 K 下,1 kPa 到100 kPa 压力区间作C2H6或C2H4在6 种高性能MOFs上的单组分吸附等温线,如图5 所示.结果表明模拟的所有吸附等温线都属于I 型曲线[49],说明是25 Å 以下微孔吸附剂.为了深入了解C2H6/C2H4吸附分离机制,在GGA-PBE/DNP水平上计算了如表1 所列的6 种高性能MOFs 的吸附能(Eads).以Al2O6-AZO_B-fum_B_No65为例,其C2H6/C2H4选择性为2.28.由吸附热结果可知,C2H6的Qst绝对值为30.09 kJ/mol,Eads为31.87 kJ/mol,分别大于C2H4的Qst绝对值(27.14 kJ/mol)和Eads值(27.96 kJ/mol).PBE 计算水平的Eads绝对值接近GCMC 水平上计算的Qst,这表明两个计算水平上的结果基本一致.因此,是热力学平衡机制导致了Al2O6-AZO_B-fum_B_No65 具有C2H6/C2H4分离选择性.但是,也正是由于吸附热之差较小,所以C2H6/C2H4选择性不大.表1 中列出的其余5 种MOFs 的选择性机理,与Al2O6-AZO_B-fum_B_No65 相同.

图5 GCMC 模拟的C2H6 和C2H4 在298 K 和1~100 kPa 条件下高性能MOFs 的吸附等温线,包括Al2O6-AZO_B-fum_B_No65,Al2O6-DPAC_B-fum_B_No152,MIL53Cr-fum_B-irmof10_B_No8,hMOF-18883,hMOF-19548 和hMOF-37529.
最后,采用MD 模拟C2H6和C2H4在上述6 种高性能MOFs 上的自扩散系数(Ds)来评估C2H6/C2H4的膜选择性,结果如表4 所示. 发现扩散选择性在0.9 附近,表明C2H4扩散比C2H6快,而膜选择性在1.83~2.08 之间.因此,上述6 个MOFs 能够用于膜选择性分离是因为C2H6/C2H4吸附选择性起主导作用.

表4 模拟的高性能MOFs 对C2H6/C2H4 的自扩散系数,吸附、扩散和膜选择性.
3.3.2 电子结构分析吸附分离机理和主客体相互作用
为深入探究选择性吸附C2H6机制,以Al2O6-AZO_B-fum_B_No65 为例,从电子结构分析了客体分子与这6 种MOFs 的相互作用,包括吸附构型、IGMH 弱相互作用分析和静电势梯度(EFGs).
首先,通过DFT 计算进一步确定气体分子的吸附位点和相互作用,结果如图6 所示.在上述6 个高性能MOFs 中,总体上C2H6比C2H4与MOFs 形成的作用位点更多而且距离更近.如图6(a)~(b)所示,C2H6与Al2O6-AZO_B-fum_B_No65 的其中一个有机配体的F 原子形成两个C-H…F 键(距离分别为3.069 Å 和3.032 Å)和与另一个有机配体的苯环形成一个C-H…键(距离为2.844 Å),而C2H4与Al2O6-AZO_B-fum_B_No65 的一个有机配体的F 原子形成两个C-H…F 键(距离分别为2.922 Å 和3.056 Å). 因此,Al2O6-AZO_B-fum_B_No65 与C2H6的作用比C2H4强,与吸附热的差值吻合.Al2O6-DPAC_B-fum_B_No152 与Al2O6-AZO_B-fum_B_No65 有相同的作用形式,如图7(a)~(b).对于MIL-53Cr-fum_B-irmof10_B_No8,C2H6和C2H4与框架均形成两个C-H…键. 但是与C2H6形成的两个C-H…键距离约为3.2 Å,而与C2H4的两个C-H…键距离分别为2.979 Å 和3.451 Å,如图7(c)~(d).采用分别去除框架上下有机配体的方式,来计算C2H6和C2H4与框架上下的C-H…作用.DFT 水平计算发现MIL-53Cr-fum_B-irmof10_B_No8与C2H6形成的两个中等强度的C-H…键,键能为15.6 kJ/mol,共31.2 kJ/mol;而C2H4形成的一强一弱的两个C-H…键,键能分别为17.3 和11.1 kJ/mol,共28.4 kJ/mol.说明MIL-53Cr-fum_B-irmof10_B_No8 与C2H6的作用更强,这与前面表1 结论一致. 如图7(e)~(f),hMOF-18883 与C2H6、C2H4均形成一个C-H…N 和一个C-H…键,且C2H6成键距离小于C2H4.如图S1(g)~(h)所示,C2H6和C2H4与hMOF-19548 分别形成两个C-H…键,且C2H6成键距离小于C2H4. 如图7(i)~(j)所示,hMOF-37529 与C2H6、C2H4均形成一个C-H…N 和一个C-H…键,且C2H6成键距离小于C2H4.
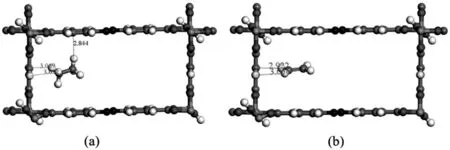
图6 优化的Al2O6-AZO_B-fum_B_No65 对(a)C2H6 和(b)C2H4 的吸附构型.
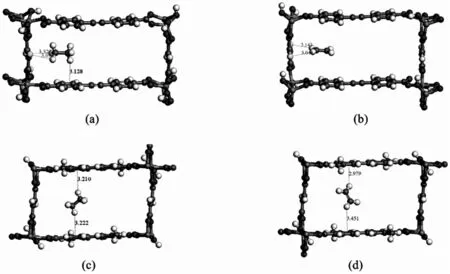
图7 优化的Al2O6-DPAC_B-fum_B_No152 对(a)C2H6 和(b)C2H4 的吸附构型、MIL53Cr-fum_B-irmof10_B_No8 对(c)C2H6 和(d)C2H4 的吸附构型、hMOF-18883 对(e)C2H6 和(f)C2H4 的吸附构型、hMOF-19548 对(g)C2H6 和(h)C2H4 的吸附构型、hMOF-37529 对(i)C2H6 和(j)C2H4 的吸附构型.
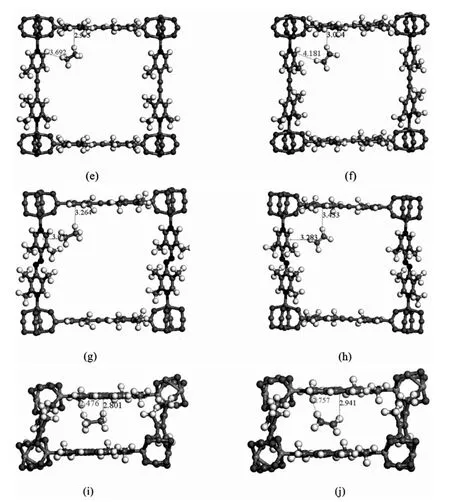
图7(续) 优化的Al2O6-DPAC_B-fum_B_No152 对(a)C2H6 和(b)C2H4 的吸附构型、MIL53Cr-fum_B-irmof10_B_No8 对(c)C2H6 和(d)C2H4 的吸附构型、hMOF-18883 对(e)C2H6 和(f)C2H4 的吸附构型、hMOF-19548 对(g)C2H6 和(h)C2H4 的吸附构型、hMOF-37529 对(i)C2H6 和(j)C2H4 的吸附构型.
其次,对DFT 水平计算的波函数进行IGMH 弱相互作用分析,计算上述6 个高性能MOFs 与C2H6、C2H4的相互作用.发现C2H6和C2H4和上述6 个MOFs 的等值面是绿色的,表明C2H6和C2H4和框架之间主要是vdW 相互作用,如图8(a)、(b)和图9 所示.而且,框架与C2H6的等值面比与C2H4的等值面大,表明C2H6和框架之间的作用力比C2H4大,与前面结合位点成键距离分析结果一致.

图8 在DFT/PBE 水平计算的Al2O6-AZO_B-fum_B_No65 对(a)C2H6、(b)C2H4 的弱相互作用IGMH 分析.

图9 在DFT/PBE 水平计算的Al2O6-DPAC_B-fum_B_No152 对(a)C2H6 和(b)C2H4、MIL53Cr-fum_B-irmof10_B_No8 对(c)C2H6 和(d)C2H4、hMOF-18883 对(e)C2H6 和(f)C2H4、hMOF-19548 对(g)C2H6 和(h)C2H4、hMOF-37529 对(i)C2H6 和(j)C2H4 的弱相互作用IGMH 分析.
最后,为了探究不同MOFs 对C2H6吸附量的差异,还计算了MOFs 的静电势梯度(EPGs). 如图10 所示,Al2O6-AZO_B-fum_B_No65、Al2O6-DPAC_B-fum_B_No152、hMOF-18883 和hMOF-19548 的静电势梯度分别为0.239、0.202、0.157 和0.142,分别对应于C2H6的吸附量1.33、1.40、1.55、1.71 mmol/g,分别为2.28、2.29、2.02和2.06.发现静电势梯度较小的MOFs 有利于C2H6的吸附.即当MOFs 的静电势梯度小,有利于C2H6吸附量的提升.这是由于C2H6的四极矩小于C2H4[50],静电势梯度小的MOF形成一个非极性的孔表面,从而有利于C2H6的吸附,因此,构建满足构效关系并且静电势梯度较小的MOFs 有利于提高C2H6的吸附量和C2H6/C2H4选择性.

图10 静电势图,包括(a)Al2O6-AZO_B-fum_B_No65,(b)Al2O6-DPAC_B-fum_B_No152,(c)MIL53Cr-fum_B-irmof10_B_No8,(d)hMOF-18883,(e)hMOF-19548 和(f)hMOF-37529.
4 结 论
本文基于结构- 吸附性能关系(SAPR),对G-MOFs 和hMOFs 数据库进行了高通量筛选吸附分离C2H6/C2H4的MOFs,并对高性能MOFs 进行机理分析.
使用构建的构效关系在G-MOFs 和hMOFs 数据库中筛选不含OMS 的高性能MOFs,分别得到2 091 和2 157 个MOFs.对于基于SAPR 筛选的4 248 个MOFs,去除含有OMS 和不含极性基团的有机配体的结构,从而有可能选择性吸附乙烷. 通过GCMC模拟验证得到6 种高性能MOFs,其C2H6吸附量超过1.05 mmol/g,C2H6/C2H4选择性超过1.99. 发现这6 种高性能MOFs 吸附等温线属于I 型曲线. 此外,MOFs 对C2H6的吸附热总体上大于C2H4的吸附热,这表明C2H6/C2H4分离是属于热力学平衡机理.此外,基于吸附位点和电子结构分析,发现C2H6与框架形成的作用位点比C2H4多且距离近.通过IGMH 弱相互作用分析,发现C2H6、C2H4与框架之间主要为vdW 相互作用.此外,根据静电势梯度计算,可知MOFs 的静电势梯度越小,越有利于C2H6吸附量的提高.这是由于C2H6的四极矩比C2H4小,易与MOFs 的非极性孔表面形成较强的相互作用.
希望本工作通过高通量计算筛选到的分离C2H6/C2H4的MOFs,为将来进一步的实验研究提供理论依据.
