Chanarin-Dorfman 综合征:1例病例报道并文献综述
2024-05-06唐建蓉
唐建蓉 贾 蓓
重庆医科大学附属第一医院感染科,重庆市传染病寄生虫病学重点实验室 (重庆, 400016)
1 病例资料
患者,男,25岁,因“全身多处鳞屑、皮肤发红20余年,瘙痒7月,加重2月”于2020年11月30日于我院就诊。患者出生时为 “火绵胶样儿”,表面薄膜剥脱后不久,皮肤开始出现红斑、鳞屑,随后诊断为先天性鱼鳞病,予以润肤治疗,皮肤情况有好转,但仍可见全身皮肤多处鳞屑,头面部、躯干皮肤发红。入院7个月前,该患者出现双侧大腿瘙痒,逐渐蔓延至全身,于我院皮肤科就诊,诊断为“疥疮”,予以硫磺软膏治疗后瘙痒有所缓解。入院前2个月因瘙痒加重,蔓延至全身,遂再次于我院皮肤科就诊,诊断为“过敏性皮炎”,予以氯雷他定片、金蝉止痒胶囊口服治疗后,皮肤瘙痒较前缓解。本次就诊时瘙痒再次加重,影响睡眠,故住院治疗。患者自发病以来无腹痛、腹胀、黄疸,无肌力异常,无听力、视力下降,无癫痫发作史。其余既往史无特殊。父母体健,否认近亲婚配,家族中无类似病史。
入院后体格检查:一般情况良好,身高165 cm,体重63 kg,体质量指数(BMI)23.1,智力、发育无明显异常。双侧睑外翻,头面部、躯干、四肢皮肤发红,面积超过90%,躯干四肢皮肤可见大量鳞屑(图1)。腹软,无压痛、反跳痛及肌紧张,肝脾肋下无触及。四肢肌力正常。
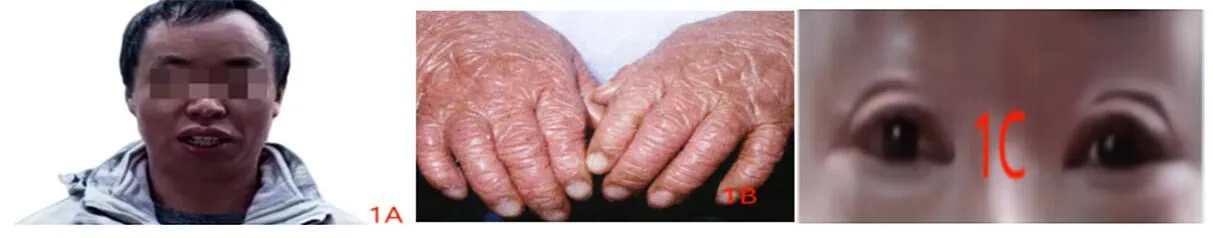
图1 患者表现为鱼鳞状红皮病、双侧睑外翻
实验室检查:丙氨酸氨基转移酶(ALT)114 U/L,天门冬氨酸氨基转移酶(AST)73 U/L,碱性磷酸酶(ALP)78 U/L,γ-谷氨酰转肽酶(GGT)71 U/L,乳酸脱氢酶(LDH)556 U/L,肌酸激酶(CK)1 186 U/L。甘油三酯、胆固醇未见升高。血沉、体液免疫、抗核抗体谱、抗中性粒细胞胞浆抗体未见明显异常。影像学检查:腹部彩超见肝脏细密,远场衰减,提示脂肪肝。心电图无明显异常。纯音测试提示患者存在双耳中度感音神经性耳聋。眼科检查提示患者存在由白内障引起的视力下降。肝脏穿刺活检(图2):脂肪性肝炎,SAF评分:S3A2F3,肝细胞脂肪变性3/3,小叶内炎症1/2,气球样变性1/2,总分5/7。纤维化分期F3。

图2 患者肝穿刺活检结果示肝细胞脂肪变性、点状坏死
考虑患者可能为Chanarin-Dorfman综合征(CDS),因此对患者进行了外显子基因检测(图3),检测到ABHD5基因的两个新的杂合突变:c.665(exon5)T>G和c.133+2(IVS2)del T(图2)。其中c.665(exon5)T>G突变可能导致外显子5的典型受体位点的消失,使得该基因序列编码的蛋白a/b水解酶结构域第222号的亮氨酸由终止密码子取代,其后的128个氨基酸均无法翻译(p.Leu222Ter,128)。c.133+2(IVS2)del T突变则为内含子突变,不造成氨基酸改变。

图3 基因分析显示两个杂合突变:c.665(exon5)T>G 和c.133+2(IVS2)del T
患者入院明确诊断为CDS,先后予以二氯醋酸二异丙胺80 mg静脉滴注1次/d,多烯磷脂酰胆碱456 mg口服3次/d,水飞蓟素140 mg口服3次/d,保肝及润肤等治疗,并采用优质蛋白、高碳水及低脂饮食。患者出院后门诊随访相关症状及肝功能等实验室指标均有所好转,近期相关复查示:ALT 91 U/L、AST 48 U/L、ALP 92 U/L、GGT 61 U/L、LDH 376 U/L、CK 680 U/L,甘油三酯、胆固醇为正常范围。患者自诉近期出现新的症状,即手指掌面和双腿处出现数枚直径4~5 mm疣状结节,平素无疼痛等,洗澡后结节稍增大,摩擦时有轻微疼痛,目前于皮肤科门诊随访中。
2 文献复习
2.1 CDS定义 CDS是一种极其罕见的、累及多系统的、常染色体隐性遗传的中性脂质体沉积病[1],最早在1953年由Jordan[2]发现,他从两个患有进行性肌萎缩症的兄弟的外周血涂片中发现了白细胞细胞质中的脂质空泡(Jordan异常)。而后Dorfman等[3]、Chanarin等[4]报道了一种中性脂质体沉积病,这种疾病的特征是脂质在外周血白细胞、骨髓粒细胞前体、肝细胞和体内其他细胞中积累。根据目前已有文献资料,迄今为止,全世界已报道的诊断该疾病的人数只有152例[5,6],我国现已报道的病例仅有5例[7-10]。该综合征由ABHD5基因突变引起,可表现为多种不同的临床症状,其中最突出的是非大疱性先天性鱼鳞样红皮病和肝脂肪变性,其他症状包括脾肿大、肌病、感音神经性耳聋、眼球震颤、共济失调、白内障、智力低下和发育迟缓等[1,11,12]。这些临床症状通常因患者的种族和不同的基因突变而表现各异。值得注意的是,所有检测到ABHD5基因突变的CDS患者均存在鱼鳞病和Jordan异常,这对CDS的诊断提供了可靠的依据。
2.2 CDS的流行病学 在全世界已报道的152例CDS患者中,男性和女性的发病率相似,但在男性中稍高,其中80例(约52.6%)为男性,72例(约47.4%)为女性[1,5,6,8]。患者年龄跨度较大,小至婴幼儿,大到中老年人均有发病。由于诊断有时会延迟,患者可能在高龄时才被诊断出来,这就导致了较高的死亡率和中老年群体发病率,随着医疗水平和科学技术的提升,该疾病的发现和诊断水平也得到提高。
地中海沿岸是CDS最常见的地区,就发生频率而言,尤以土耳其最高,印度其次,中东地区第三。全世界CDS患者中有近一半的父母是近亲,土耳其、印度和中东等国家或区域的近亲结婚率高于世界平均水平,因此,这些地区的纯合子和杂合子患者比例高于其他地区[1]。而在中国目前所报道的病例极少,暂时未发现明显的地区及种族差异。
2.3 CDS的临床表现 由于CDS是累及多系统的复杂疾病,其临床表现多种多样,但通常以皮肤病变起病,非大疱性鱼鳞状红皮病是CDS患者的特征性表现[11],主要表现为皮肤表面出现红斑且伴白色鳞屑。在目前所发现的病例中,所有患者均有鱼鳞病的主要症状。
肝脏是CDS最常见的受累器官之一,患者常表现为肝肿大、脂肪肝和肝酶升高。据报道,约85%的患者可见肝脏受累,最常见的病理表现为脂肪肝,可进一步发展为肝纤维化和肝硬化[1]。目前已有文献报道中,所有的CDS患者均未被诊断为肝细胞癌。一些患者可有脾肿大,多通过体格检查和腹部B超检查发现。Mitra等[11]曾报道过1例20月大的男性CDS患儿,其腹部检查发现10 cm坚硬、无触痛的肝肿大伴轻度脾肿大,且存在双侧隐睾症。但隐睾症是否与CDS相关联目前暂不明确。
肌病是CDS的另一常见表现,由于ABDH5突变导致ATGL脂解效率降低,脂滴在肌纤维中积累,降低了骨骼肌中的肉碱水平。此外,由于这些脂滴通常用于磷脂的合成,它们的缺乏会导致磷脂膜的异常,从而导致肌病的发展。可通过体格检查、CK水平和肌肉活检进行诊断。患者可表现为肌无力、心肌病等。据报道,约60%的患者存在CK升高及肌无力[1]。Arora等[13]报道患者肌电图检查显示早期复张,具有提示肌病的完全间期模式。这提示我们还可通过肌电图来明确诊断。
一些文献报道中CDS患者有共济失调、智力低下和小头畸形[12],以及感音神经性耳聋、眼球震颤、睑外翻和斜视、白内障和生长迟缓等常见表现,患者通常可经过相应专科检查发现。此外,还有一系列少见甚至罕见的临床表现,如精神疾病、血管瘤、纵隔神经肿瘤、小耳、佝偻病、肾脏受累和内分泌腺空泡[11,14,15]。
不可否认的是,由于该病较罕见,且表现多样,许多患者或许未能明确诊断,亦或缺乏详细记录,CDS可能还存在其他相关临床表现,有待进一步发掘。
2.4 CDS的发病机制 CDS是由位于3p21染色体上的ABHD5基因突变引起的,该基因又称为CGI-58基因。该基因的产物是α/β水解酶蛋白家族的成员,含有349个氨基酸,分子质量约39 kD,能够激活在细胞内脂肪分解中发挥重要作用的脂肪甘油三酯脂肪酶(ATGL)。ABHD5基因突变导致ATGL的全部或部分失活,从而使脂肪酸从脂肪细胞和非脂肪组织细胞内三酰甘油存储中释放出来,这导致细胞内脂肪酸动员不足,脂滴在肝细胞、肠黏膜、血液、骨髓、皮肤、纤维细胞、肌细胞、中枢神经系统细胞和许多其他类型的细胞中堆积[16,17 ],产生各系统不同的临床表现。患者外周血涂片可以观察到中性粒细胞内的特征性脂滴,称Jordan畸形,这也是CDS的主要诊断标准之一。
鱼鳞病产生与皮肤通透性屏障缺陷有关。脂类具有高疏水性,于渗透性屏障的形成至关重要。在正常的皮肤中,颗粒层和角质层之间的间隙充满了脂质层膜,这些脂质层主要由神经酰胺、胆固醇和游离脂肪酸组成。由此可见表皮中的酰基神经酰胺是影响皮肤渗透性屏障的重要因素。含有PNPLA1酶的蛋白类磷脂酶结构域在酰基神经酰胺的合成中起着关键作用。ABHD5基因可激活ATGL,而ATGL实际上属于PNPLA家族,也被称为PNPLA2。PNPLA1和PNPLA2之间存在序列的相似性[1]。Yusuke等[18]研究发现,当ABHD5在产生酰基神经酰胺的细胞系统中表达时,PNPLA1的酰基神经酰胺合成得到增强。因此,在CDS中发现的ABHD5基因突变降低了ABHD5基因促进PNPLA1依赖性酰基神经酰胺产生的能力,导致皮肤屏障功能受损,从而产生鱼鳞状红皮病等表现。然而,尚未发现CDS的肝脏和神经系统等症状与ABHD5和PNPLA1或PNPLA2之间相互作用的关系。这些症状可能是ABHD5与PNPLA家族的其他蛋白相互作用的结果。
2.5 CDS的诊断 CDS主要通过患者特征性临床表现及外周血涂片镜检发现Jordan畸形来明确诊断。在极少数情况下,在粒细胞中没有发现脂滴时,需要对其他组织样本进行检查,如果怀疑指数很高,则需要使用电子显微镜。临床中常使用超声和病理检查等明确肝脏病变情况。此外,近年来测序技术随着高通量二代测序的出现得到进一步发展,因此可作为另一种诊断方法,还可对ABDH5基因突变进行遗传分析。
2.6 ABHD5基因突变的类型 ABHD5基因的突变方式包括剪接位点突变、插入突变、缺失突变、无义突变和错义突变。患者中最常见的突变类型是c.594insC p.N209X,其他突变大多是家族性或局部性的。目前相关文献中有明确记录的CDS患者基因突变情况见表1。

表1 CDS基因临床特征及突变类型[1,5,6,8]
2.7 CDS的治疗 CDS目前还没有确切有效的治疗方法,据报道,高碳水化合物和含有中链甘油三酯(MCTs)的低脂饮食可以改善患者皮肤和肝脏的症状[19]。对于严重鱼鳞病患者,维甲酸(如阿维A)是首选治疗方法,但是患者存在肝功能受损时不能给予维甲酸。同时局部涂抹润肤剂的治疗是有效的[20]。AlNeyadi等[5]报告的病例中提到,低脂肪酸饮食以及中链甘油三酯补充剂可能会减少肝肿大并使肝酶降至正常范围,特别是早期开始并与维生素E(10 mg/kg·d)和熊去氧胆酸(UDCA,15~20 mg/kg·d)结合使用时。Cakmak等[1]也推荐CDS患者食用富含碳水化合物、中链脂肪酸和UDCA的饮食,以及局部应用含尿素的润肤剂。
对于晚期肝硬化患者,应给予普萘洛尔、螺内酯、利尿剂等对症治疗,并建议进行肝移植。肝移植可以作为失代偿期肝硬化的CDS患者的一种选择,但是,这类患者仍需要饮食疗法和长期监测。
总之,CDS是一种罕见的遗传性疾病,临床表现多种多样,早期多以皮肤病变起病,因此容易漏诊和误诊。在临床诊疗中,对于先天性鱼鳞病患者同时存在其他多系统表现,如听力下降、肝肿大、脂肪肝、白内障、角膜病变、肌病和智力低下时,需考虑存在CDS可能,并可尽早行外周血涂片镜检诊断,同时行ABHD5基因检测进一步诊断,从而早期开始针对性治疗,以期改善患者生活质量。基因检测有助于确定ABDH5基因的突变类型,进而分析基因型与表型的相关性,基因分析也可能对解释CDS患者的临床表现和预测预后较为重要。CDS的治疗目前大多只是对症治疗,未来或许能通过收集更多CDS患者的临床特征及基因分析,为进一步研究疾病的发生发展和治疗奠定基础。
注:本文病例报道中相关信息已获得患者本人认可及同意公开发表。感谢患者对我们研究工作的积极无私大力支持!
