腹腔巨大间质肿瘤伴小肠间质肿瘤1例
2024-04-25李长平郭吉庆
李长平,郭吉庆
(通化市人民医院,吉林 通化 134001)
1 病历摘要
患者女,67岁。因发现腹部巨大肿物半年于2022年9月13日入院。无触痛,一般状况尚可,营养良好。体检:左侧腹部可触及一巨大包块,约“儿头”大小,质地硬,无触痛,活动度差,表面凸凹不平。腹部及盆腔CT:全腹(肠道)(平扫+增强扫描):腹腔内巨大肿瘤;肝右前叶、右后叶分别由肝固有动脉及肠系膜上动脉分支供血,先天变异;右侧壶腹型肾盂,左肾小囊肿。行肿瘤及一段小肠切除术。腹腔无腹水。病理检查:巨大肿瘤1枚,大小:25 cm×25 cm×12 cm,被膜完整,表面覆脂肪组织;切面灰白,质嫩,鱼肉状,伴囊性变,坏死,略带黏液感。小肠一段,长约5.5 cm,直径1.5 cm,小肠系膜对侧表面见肿瘤1枚,大小:7.0 cm×7.0 cm×4.0 cm;切面灰白,质嫩,出血坏死,肿瘤贯穿肠壁突出于肠管内,肠管内肿瘤部分直径1.3 cm,小肠黏膜皱襞存在,壁厚0.5 cm。镜下观察:腹腔肿瘤与小肠肿瘤组织学相似,梭形细胞为主,呈交织的短条束状或漩涡状排列,部分区域呈长条束状或鱼骨样排列(图1),局部出血坏死,细胞中等异型性,核分裂像>10个/50 HP(图1),肿瘤侵犯神经组织,未见明确脉管侵犯。其中小肠肿瘤向内凸向肠腔,侵犯黏膜下层、肌层,肿瘤主体位于肠管外。免疫组化相同项目有:CK(-)、DOG1(+)(图2)、CD117(+)(图2)、SMA(-)、DES(-)、S-100(-)、KI67约2%~3%(+);CD34(-)。病理诊断:(腹腔内及小肠)间质肿瘤,高危险度。随访1个月,患者无明显不适,口服伊马替尼。本研究经过本院医学伦理委员会同意。
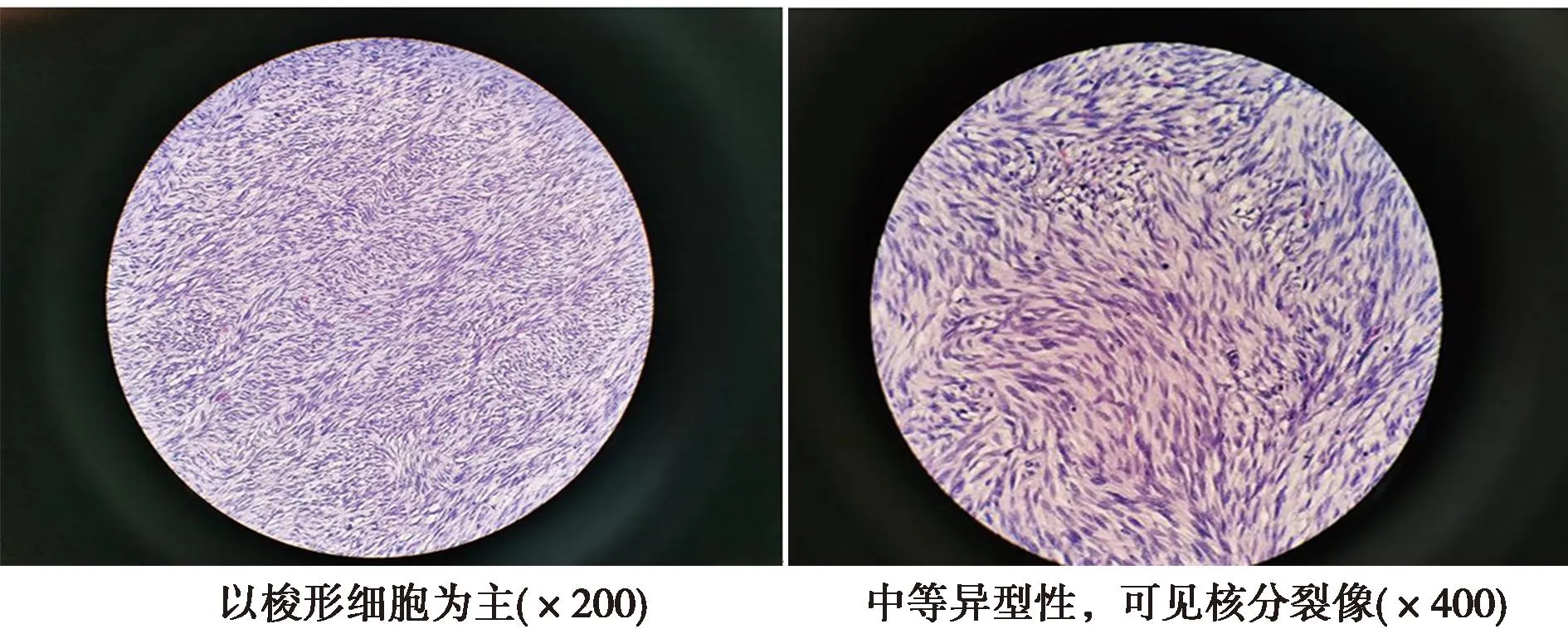
图1 病理组织学观察(HE)
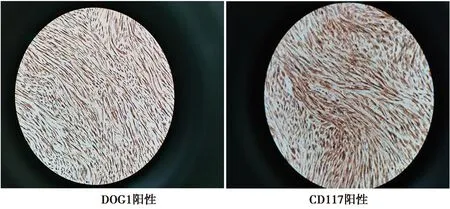
图2 肿瘤组织免疫组化染色(DAB,×200)
2 讨论
胃肠道间质肿瘤(GIST)是胃肠道常见的间源性肿瘤。好发于成年人(>50岁),儿童罕见。大多数的GIST发生于胃(60%),部分病例发生于小肠(30%),少数病例还可发生于结直肠、阑尾和食道及胃肠道外,后者包括大网膜、肠系膜、盆腔或腹膜后。临床表现缺乏特异性,晚期多表现为消化道出血及腹部肿块,早期无任何症状。
GIST命名由Mazur和CLark[1]于1983年首次提出。GIST中的瘤细胞起自于卡哈尔间质细胞或向卡哈尔间质细胞分化的未定型细胞。目前将GIST定义为肿瘤细胞表达c-kit(CD117)或血小板转化生长因子(PDGFRA)功能获得性突变的非上皮性(间叶性)肿瘤[2]。约15%成人和>90%的儿童胃肠道间质肿瘤属于野生型)无c-kit或PDGFRA突变),其中约42%的野生型GIST表现为琥珀酸脱氢酶亚单位(SDH)基因缺失突变,免疫表型呈SDHB表达缺失。
GIST可位于黏膜下、肌层内或浆膜下,外观呈结节状,边界相对清楚,直径可数毫米至数十厘米,中位直径6.0 cm,切面灰白或灰红色,质嫩,可见出血、坏死及囊性变。发生于腹腔内者可有纤维性假包膜。本例发生于腹腔内(较大肿瘤),也称为胃肠道外间质肿瘤(E-GIST)[3-6]。组织学形态主要分为4种类型(根据2022版CSCO胃肠间质瘤诊疗指南):梭形细胞为主型(50%~70%)、上皮样细胞为主型(20%~40%)、混合细胞型(10%)及去分化GIST(新增类型)。梭形细胞为主型GIST中瘤细胞多呈条束状或编织状排列(如本例镜下表现即是),细胞呈梭形或短梭型,局部排列稀疏,局部密集,且核分裂易见,细胞核异型性轻重不一。上皮样细胞为主型GIST瘤细胞似浆细胞样,此病例本形态不典型。去分化GIST包括原发性(肿瘤内出现非GIST高级别肉瘤,如未分化肉瘤等)和靶向治疗后(如出现横纹面肉瘤等高级别肉瘤转化)。肿瘤间质改变:胶原化或玻璃样变,明显时可硬化性改变,小肠GIST 中常可见丝团样纤维;钙化或骨化;黏液样变;较多的炎性细胞浸润;出血和囊性变;坏死,并常见瘤细胞围绕血管生长。免疫组化:大部分GIST表达CD117,多呈胞膜和(或)胞质弥漫强阳性。DOG1敏感性与CD117几乎相同,与CD117联合用于GIST的诊断具有较好的互补性。此外部分GIST还可表达CD34(占70%~80%);SMA、calponin、h-caldesmon(占30%~40%);desmin、s-100(<5%)。2022版CSCO胃肠间质瘤诊疗指南将CD117、DOG1、KI67、SDHB(胃)作为Ⅰ级推荐,另外常规加用ki-67和CD34标记。PDGFRA标记对预示PDGFRA基因突变有潜在性价值,但最终诊断仍需分子检测明确。
靶向治疗后的病理形态和病理学疗效评估:大体显示退行性改变,质地和颜色与经典GIST不同,包括出血、坏死和囊性变等。镜下表现瘤细胞稀疏,间质常伴有广泛胶原化,可伴有组织细胞反应、含铁血黄素沉着、炎性细胞浸润、出血和囊性变等。部分病例可有异质性。GIST术前靶向治疗后的病理学效应评估(试用):①完全效应:无瘤细胞残留;②高度效应:稀疏瘤细胞残留(残留瘤细胞≤5%;③部分效应:明显瘤细胞残留(残留瘤细胞>5%~95%),但可见间质胶原化、组织细胞反应、炎性细胞浸润、含铁血黄素沉着和坏死等改变。④零级效应:瘤细胞和间质均无相应变化(残留瘤细胞>95%)。
GIST分子病理检测:适应人群包括:常规病理不易诊断者;活检病理诊断为GIST,术前拟行靶向治疗;术后病理评估为中-高危险度,拟行靶向治疗者;继发耐药性GIST;复发性或转移性GIST。分子检测内容包括:常规检测;继发突变肿瘤;野生型GIST。KIT突变:与第9、11、13、17号外显子和PDGFRA基因第12、14、和18号外显子相关。PDGFRA突变较少见;SDH缺陷型GIST对甲磺酸伊马替尼反应差。野生型GIST:是指病理诊断符合GIST但分子检测无KIT/PDGFRA基因突变者,大多数见于儿童及少数成人,分为SDH缺陷型GIST及非SDH缺陷型GIST。家族性GIST多为常染色体显性遗传,占比不足5%,主要表现为胃肠道多发性GIST,对存在胚系突变的患者亲属,应早发现、早诊断和早治疗。
GIST鉴别诊断:①胃肠道平滑肌瘤:瘤细胞的排列稀疏,细胞之间的间距比较宽,瘤细胞的胞质呈深嗜伊红色,细胞无异型性,核长梭型,两端钝圆,不见核分裂像,免疫组化标记显示瘤细胞表达SMA和Des,不表达CD117。②肠系膜或盆腔内韧带样纤维瘤病:由梭形纤维母细胞或肌纤维母细胞组成,常呈平行状或长的条束状排列,间质可伴有粘液变性,瘤细胞表达β-catenin核着色,一般不表达CD117。③胃肠道型神经鞘瘤:肿瘤一般<5 cm,其镜下特点为:多数病例于肿瘤的周围可见淋巴细胞组成的淋巴细胞套,肿瘤细胞可有致密区及疏松区,有时可见栅栏状排列,免疫组化显示S-100蛋白、GFAP阳性,不表达CD117。④胃肠道血管球瘤:瘤细胞圆形,大小一致,胞质淡染,界清,瘤细胞表达SMA、actin,不表达CD34。⑤腹膜后平滑肌肉瘤:组织学形态与GIST有交叉,免疫组化表达SMA、H-caldesmon,不表达CD117和CD34。⑥胃肠道透明细胞肉瘤:瘤细胞表达S-100蛋白、HMB45,可有EWS-ATF1融合蛋白。⑦胃肠道卡波西肉瘤:部分患者有肝、肾移植,瘤细胞表达CD34和VEGF3R。⑧腹腔内结外滤泡树突细胞肉瘤:肿瘤显示双相细胞性形态,即由胖梭形、卵圆形至上皮样的瘤细胞和多少不等的小淋巴细胞形成。瘤细胞多呈席纹状、漩涡状或条束状排列,也可呈片状分布;淋巴细胞夹杂于肿瘤内或围绕在血管周围,形成血管淋巴套。瘤细胞表达CD19、CD21、CD23、CD35,不表达CD34、CD117。
GIST危险度分为极低、低、中等、高四个级别。预后与危险度分级密切相关,与肿瘤生长方式、肿瘤的大小、肿瘤破裂、胃肠道穿孔、有无腹水、有无黏膜侵犯、血管浸润、神经浸润、脂肪浸润、黏膜浸润、肌层浸润、围绕血管呈周皮细胞瘤样、有无邻近器官侵犯、细胞密度、细胞的异型性、核分裂像的多少、有无凝固性坏死和有无远处转移等因素有关。
综上,GIST诊断并不难,是消化道最常见的间叶源性肿瘤,只要恰当应用免疫组化及分子检测(KIT/PDGFRA基因突变)可避免漏诊,靶向药甲磺酸伊马替尼(格列卫,酪氨酸激酶抑制剂)对GIST有显著疗效,准确、规范的病理诊断对GIST的临床治疗和预后判断至关重要。
