N6-甲基腺苷修饰在肿瘤程序性细胞死亡中的作用*
2024-04-24谈元郡王霞黄静综述张百红审校
谈元郡 王霞 黄静 综述 张百红 审校
肿瘤细胞的关键特征之一为非突变表观遗传重编程,是动态转录组异质性的驱动力。在人类肿瘤中,表观遗传学改变如DNA 甲基化、组蛋白修饰、RNA修饰、微小RNA 和核小体重塑等均控制机体关键基因及蛋白表达,调节机体的生理或病理过程。N6-甲基腺苷修饰(N6-methyladenosine,m6A)是真核生物体内最丰富的转录后表观遗传修饰方式,mRNA 在m6A甲基转移酶作用下发生腺苷酸第六位N 原子甲基化,甲基化腺苷也可在m6A 去甲基化酶作用下还原为腺苷,m6A 结合蛋白识别经m6A 修饰的mRNA 并调节mRNA 的可变性剪接、衰变和翻译等,进而调节机体一系列生理及病理过程。
抵抗程序性细胞死亡是肿瘤另一关键特征,抵抗程序性细胞死亡使得肿瘤细胞获得持续恶性增殖的能力。不同于物理、化学或生物损伤导致的非程序性细胞坏死,程序性细胞死亡依赖于复杂的分子调控机制。研究发现,m6A 修饰参与凋亡、自噬、焦亡、铁死亡、坏死性凋亡、铜死亡等肿瘤程序性细胞死亡形式的调节。本文就m6A 修饰对常见的肿瘤程序性细胞死亡方式的调节进行阐述。了解肿瘤细胞程序性死亡的调节机制有助于进一步阐明肿瘤进展的机制,也可为临床肿瘤治疗提供新的策略。
1 m6A 修饰与肿瘤细胞凋亡
凋亡是细胞在接触生理性或病理性刺激后为维持内环境稳定而发生的程序性细胞死亡过程,主要是由内在线粒体外膜透化启动或外在细胞表面死亡受体与其死亡诱导配体结合激活caspase 蛋白酶触发。目前研究表明m6A 修饰通过抑制或促进肿瘤细胞凋亡在肿瘤细胞凋亡调控过程中发挥双重作用。
1.1 抑制肿瘤细胞凋亡
Wang 等[1]发现m6A 甲基转移酶甲基转移酶样3(methyltransferase-like protein 3,METTL3)介导胃癌细胞中长链非编码RNA ABL 的m6A 修饰,维持ABL稳定性。ABL 与凋亡蛋白酶激活因子1 的WD1/WD2结构域结合竞争性地阻止细胞色素C 与凋亡蛋白酶激活因子1 的相互作用,阻断细胞凋亡体的组装和caspase 3/9 的激活,抑制肿瘤细胞凋亡。m6A 甲基转移酶RNA 结合基序蛋白15(RNA binding motif protein 15,RBM15)介导的m6A 修饰上调宫颈癌中OTU 结构域的泛素醛结合蛋白2(OTU domain-containing ubiquitin aldehyde-binding protein 2,OTUB2)表达,高表达的OTUB2 通过刺激AKT/mTOR 信号通路抑制肿瘤细胞凋亡[2]。m6A 去甲基化酶脂肪量和肥胖相关蛋白(fat mass and obesity-associated protein,FTO)介导乳腺癌细胞中Bcl-2 家族促凋亡蛋白Bcl-2/腺病毒E1B19kD 相互作用蛋白3(Bcl-2/adenovirus E1B-19 kDa-interacting protein 3,BNIP3)mRNA的m6A 甲基化腺苷去甲基化,并诱导BNIP3 mRNA降解,下调BNIP3 的表达进而抑制肿瘤细胞凋亡[3]。FTO 介导的m6A 去甲基化降低结直肠癌中凋亡诱导因子SIVA1 的表达,下调SIVA1 对caspase3 的激活作用,进而抑制肿瘤细胞凋亡,促进肿瘤进展[4]。m6A去甲基化酶AlkB 同源蛋白5(Alk B homologue 5,ALKBH5)介导的m6A 去甲基化增强了食管鳞状细胞癌中长链非编码RNA CASC8 转录本的稳定性,上调CASC8 表达,高表达的CASC8 与核不均一核糖核蛋白L(heterogeneous nuclear ribonucleoprotein L,HNRNPL)相互作用激活Bcl-2/caspase3 通路,抑制肿瘤细胞凋亡,促进食管鳞状细胞癌进展[5]。在急性髓系白血病中ALKBH5 介导的去甲基化降低了mTOR 相关蛋白MLS8/真核翻译起始因子4E 结合蛋白1(eukaryotic translation initiation factor 4E-binding protein 1,EIF4EBP1)mRNA 的m6A 修饰水平,导致转录本稳定性下降,下调caspase3/7 活性,抑制肿瘤细胞凋亡[6]。逆转凋亡抑制信号的异常m6A 修饰可促进肿瘤细胞凋亡,抑制肿瘤进展。Zhang 等[7]发现龙胆草75%的乙醇提取物通过下调METTL3 的表达降低METTL3与抗凋亡蛋白Survivin mRNA 的结合率,抑制Survivin mRNA 中的m6A 修饰,下调抗凋亡蛋白Survivin 的表达,进而诱导caspase3 和Poly 聚合酶等凋亡执行蛋白的裂解激活,诱导肿瘤细胞凋亡。此外,也有研究发现二甲双胍可通过抑制多发骨髓瘤中METTL3 介导的m6A 修饰来抑制USP4 的表达,从而促进肿瘤细胞凋亡,阻碍细胞恶性增殖[8]。
1.2 促进肿瘤细胞凋亡
Li 等[9]发现METTL3 介导膀胱癌中长链非编码RNA LNPPS 的m6A 修饰,增加LNPPS 的稳定性,上调其表达,高表达的LNPPS 作为膀胱癌中凋亡促进信号PDCD5 和P53 的支架,阻断PDCD5 K20 位点泛素化并破坏双微体2 介导的P53 泛素化,增强与P53 相关的细胞凋亡信号,促进肿瘤细胞凋亡。雌激素受体阳性HER2 阴性乳腺癌细胞中METTL3 以m6A 依赖性方式促进caspase 蛋白酶的上游因子BAX 表达,进而上调caspase 家族活性,促进肿瘤细胞凋亡[10]。三阴性乳腺癌中YT521-B 同源性域家族2(YT521-B homology domain family proteins 2,YTHDF2)与丝裂原活化蛋白激酶MAPK 途径中编码蛋白的mRNA 相互作用稳定MAPK 途径中的靶标mRNA,促进未折叠蛋白积累,进而引起内质网应激介导的细胞凋亡[11]。ALKBH5 通过降低胰腺癌中CUGBP Elav 样家族成员2(CUGBP Elav-like family member 2,CELF2)的m6A 修饰水平上调CELF2 的表达,进而抑制Bcl-2 家族抗凋亡蛋白Mcl-1 表达,促进肿瘤细胞凋亡,抑制胰腺癌恶性进展[12]。逆转凋亡促进信号的异常m6A 修饰也可促进肿瘤细胞凋亡,抑制肿瘤进展。Zhang 等[6]发现生物活性肽通过下调ALKBH5 抑制mTOR 相关蛋白MLS8/EIF4EBP1 mRNA的m6A 去甲基化,促进EIF4EBP1 表达及其促凋亡活性发挥抗肿瘤作用(表1)。
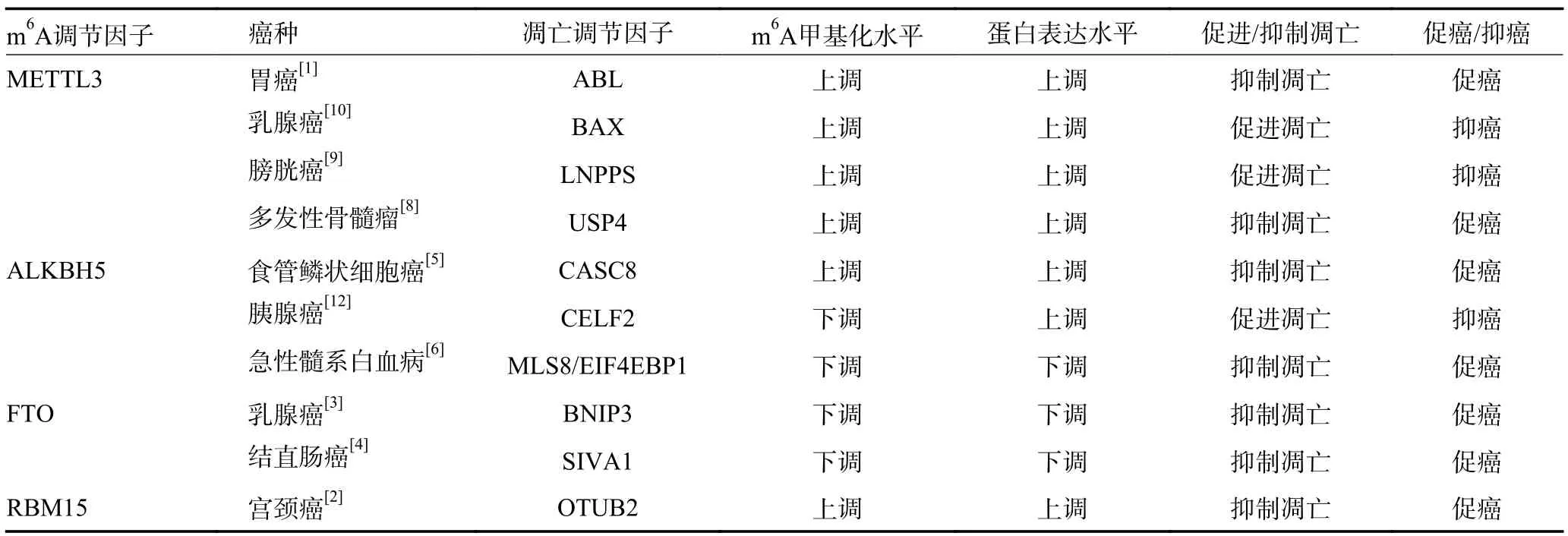
表1 肿瘤细胞凋亡中的m6A 甲基化调控
2 m6A 修饰与肿瘤细胞自噬
自噬是一种细胞内自我降解机制,在肿瘤发展的不同阶段起着动态的肿瘤抑制或促进作用。早期肿瘤细胞自噬可清除受损的蛋白质和细胞器,维持基因组的稳定性,抑制肿瘤进展。中晚期的肿瘤细胞因受到环境压力的影响,将自噬作为一种细胞保护机制,以维持肿瘤细胞线粒体的功能,减少DNA 损伤,提高肿瘤细胞的存活率和抗应激(如营养剥夺、缺氧等)能力,促进肿瘤进展。机体对细胞自噬的调控涉及多种自噬相关蛋白和通路,研究表明,m6A 甲基化修饰介导这些关键蛋白和信号通路传导因子的异常表达在肿瘤细胞自噬中发挥重要作用。
2.1 抑制肿瘤细胞自噬
2.1.1 抑制mTOR 通路依赖性肿瘤细胞自噬 自噬分为自噬启动、自噬膜延伸和自噬溶酶体形成三个过程。自噬启动的标志是自噬体的形成,启动过程依赖于UNC-5 类似自噬激活激酶(UNC-5 like autophagy activating kinase,ULK)复合物,ULK 复合物上游信号通路主要涉及mTOR 依赖性途径和非mTOR 依赖性途径。mTOR 通过磷酸化包括自噬相关蛋白13(autophagy-related 13,ATG13)和ULK1/2 在内的复合物成分,抑制ULK 复合物介导的自噬启动过程。此外,mTOR 也可通过抑制Beclin1 调节因子1 的磷酸化抑制ULK 稳定性。m6A 修饰通过调节mTOR 介导的自噬抑制信号促进多种肿瘤进展。非小细胞肺癌中ALKBH5 的上调使癌基因泛素结合酶E2C(ubiquitinconjugating enzyme E2C,UBE2C)mRNA 维持在较低的m6A 修饰水平,增加了UBE2C mRNA 的稳定性,上调UBE2C 在非小细胞肺癌中的表达,高表达的UBE2C 促进含DEP 结构域的mTOR 相互作用蛋白DEPTOR 的泛素化和降解,从而促进mTOR 信号的激活,进而选择性抑制非小细胞肺癌自噬,诱导肿瘤细胞侵袭性生长[13]。上皮性卵巢癌组织中过表达的ALKBH5 介导的m6A 修饰去甲基化激活EGFR/PIK3CA/AKT/mTOR 信号通路抑制上皮性卵巢癌细胞自噬,促进肿瘤细胞增殖[14]。m6A 识别蛋白胰岛素样生长因子2 信使RNA 结合蛋白3(insulin-like growth factor 2 mRNA binding protein 3,IGF2BP3)结合喉鳞状细胞癌中经m6A 修饰的机械翻译相关蛋白同源物7(translation machinery associated 7 homolog,TMA7)并促进TMA7 的表达,TMA7 上调通过激活P13K/mTOR 通路抑制肿瘤细胞自噬,促进喉鳞状细胞癌进展[15]。此外,METTL3 以m6A 依赖性方式促进急性髓系白血病长链非编码RNA PSMA3 反义RNA1 表达,进而激活mTOR 信号上游P13K/AKT 通路,抑制肿瘤细胞自噬,促进急性髓系白血病进展;介导慢性粒细胞白血病中PTEN mRNA 的m6A 修饰,降低PTEN稳定性,增加mTOR 上游信号通路P13K/AKT 活性,抑制肿瘤细胞自噬并促进慢性粒细胞白血病进展[16]。
2.1.2 抑制非mTOR 通路依赖性肿瘤细胞自噬 近年研究发现m6A 修饰不仅参与了mTOR 通路依赖性自噬抑制信号,在独立于mTOR 通路的自噬抑制信号中同样发挥作用。m6A 甲基转移酶Wilms 瘤1-相关蛋白(Wilms' tumor 1-associating protein,WTAP)在肝细胞癌中介导的m6A 修饰降低肝激酶基因B1(liver kinase B1,LKB1)mRNA 的稳定性,下调LKB1 表达,抑制AMP 活化蛋白激酶磷酸化水平进而下调肝细胞癌的AMP 激活蛋白激酶信号传导,抑制肿瘤细胞自噬,促进肝细胞癌进展[17]。FTO 介导的m6A 修饰去甲基化在口腔鳞状细胞癌中通过靶向真核翻译起始因子4G1(eukaryotic translation initiation factor 4 gamma 1,eIF4G1)的转录本,上调eIF4G1 表达,抑制肿瘤细胞自噬,促进口腔鳞状细胞癌进展。敲低口腔鳞状细胞癌细胞系中FTO 的表达后eIF4G1 下调,肿瘤细胞自噬通量增强[18]。乳腺癌细胞中,eIF4G1 表达也因FTO 的m6A 去甲基化酶活性上调,高表达的eIF4G1 抑制了乳腺癌细胞自噬,促进乳腺癌细胞增殖和转移[19]。盐诱导激酶2(salt-inducible kinase 2,SIK2)属于AMP 激活蛋白激酶家族成员,通过磷酸化Ser317 和Ser777 直接激活ULK1,促进自噬体加工成熟,FTO 通过m6A 依赖性方式降低肾透明细胞癌中SIK2 mRNA 的稳定性,抑制肿瘤细胞自噬,促进肿瘤进展[20]。此外,m6A 结合蛋白核不均一核糖核蛋白A2/B1(heterogeneous nuclear ribonucleoproteins A2-B1,HNRNPA2B1)通过识别下游自噬相关蛋白ATG4B 转录本3’UTR 中的m6A 位点促进ATG4B m-RNA 衰变,继而下调乳腺癌细胞系的自噬通量,促进恶性细胞增殖[21]。
2.2 促进肿瘤细胞自噬
2.2.1 促进mTOR 通路依赖性肿瘤细胞自噬 DNA损伤诱导转录因子4(DNA damage inducible transcript 4,DDIT4)通过激活TSC1/2 复合物抑制mTOR 信号通路激活自噬,ALKBH5 介导头颈部鳞状细胞癌中DDIT4 的m6A 修饰去甲基化,降低DDIT4 转录本m6A 水平并提高DDIT4 mRNA 的稳定性,促进肿瘤细胞自噬,促进肿瘤进展[22]。FTO 介导结直肠癌中转录激活因子4(activating transcription factor 4,ATF4)mRNA 的m6A 去甲基化,降低ATF4 转录本的m6A 修饰水平,阻止ATF4 在结肠癌细胞中的降解延长半衰期,高表达的ATF4 激活DDIT4 的转录使mTOR 信号失活,诱导利于肿瘤细胞生存的自噬过程,促进肿瘤恶性进展[23]。
2.2.2 促进非mTOR 通路依赖性肿瘤细胞自噬 RB1-诱导卷曲蛋白1(RB1-inducible coiled-coil 1,RB1CC1)和ULK1 为自噬启动复合物的组成亚基,METTL14以m6A 依赖性方式促进口腔鳞状细胞癌自噬相关基因RB1CC1 的表达,促进自噬溶酶体形成,提高自噬通量,抑制口腔鳞状细胞癌细胞增殖[24]。METTL3 以m6A 依赖性方式增强泛素特异性肽酶13(ubiquitin specific protease,USP13)mRNA 的稳定性,USP13 通过去泛素化稳定ATG5,进而促进胃肠道间质瘤细胞自噬以及肿瘤细胞对伊马替尼的抗性,促进肿瘤进展[25]。非小细胞肺癌中高表达的FTO 通过降低生长停滞特异性转录本5(growth arrest-specific transcript 5,GAS5)m6A 甲基化水平抑制GAS5 的表达,促进非小细胞肺癌的自噬性死亡,抑制肿瘤进展[26]。顺铂耐药胃癌细胞中FTO 介导的m6A 去甲基化上调顺铂耐药胃癌细胞中ULK1 的表达,ULK1 通过募集其他自噬相关蛋白启动自噬体形成,促进细胞自噬及自噬诱导的顺铂耐药,FTO 敲低后ULK1 mRNA 水平显著下调,FTO 沉默可以通过灭活体内ULK1 依赖性自噬来增加胃癌细胞对顺铂的敏感性[27]。缺氧诱导因子-1α 在缺氧状态下通过直接结合m6A 结合蛋白YTH家族蛋白1(YTH domain family protein 1,YTHDF1)启动子区域调节YTHDF1 转录,进而促进YTHDF1与m6A 修饰的ATG2A 和ATG14 mRNA 结合,促进ATG2A 和ATG14 的翻译继而促进肝细胞癌缺氧诱导的自噬,促进肿瘤进展[28](表2)。
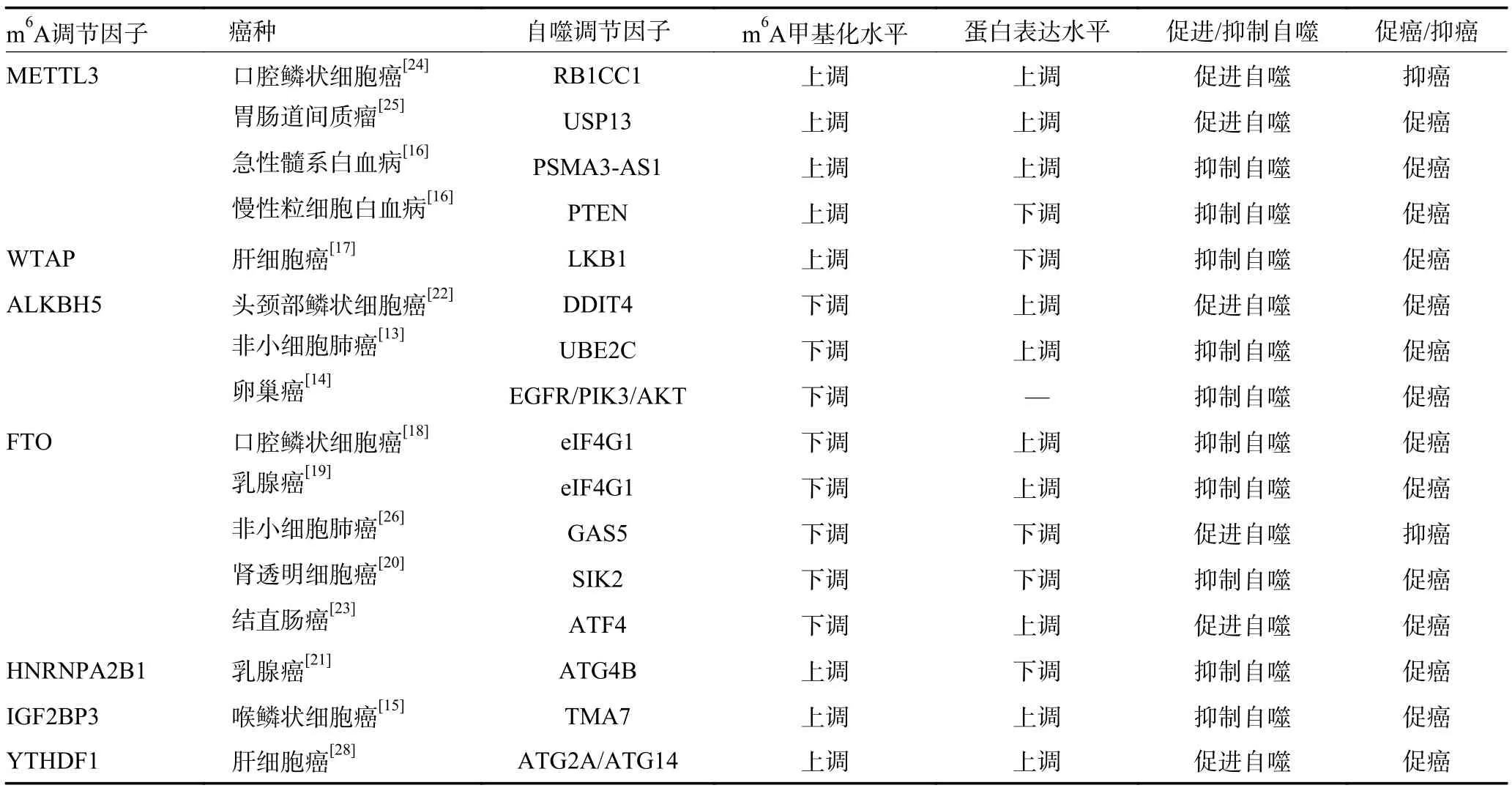
表2 肿瘤细胞自噬中的m6A 甲基化调控
3 m6A 修饰与肿瘤细胞焦亡
焦亡是一种新发现的程序性细胞死亡形式,Nod样受体蛋白3(Nod-like receptor protein 3,NLRP3)等炎性小体的激活诱导caspase1 裂解并分离Gasdermin D(GSDMD)的N 端和C 端以触发经典的焦亡途径。此外,caspase4/5/11 也可通过非炎性小体途径直接识别细胞内LPS 受体并与之结合介导GSDMD 切割,诱导细胞焦亡。焦亡在肿瘤发展过程中发挥双重作用,肿瘤中央缺氧区肿瘤细胞焦亡引起的慢性肿瘤细胞坏死抑制了机体抗肿瘤免疫,加速肿瘤进展,而肿瘤微环境中细胞焦亡诱导的急性炎症增强免疫反应并抑制肿瘤的进展。目前研究表明m6A 修饰参与了肿瘤细胞经典焦亡途径(表3)。在下咽鳞状细胞癌中METTL3 介导环状RNACUX1 的m6A 修饰并稳定其表达,环状RNACUX1 与caspase1 结合并抑制其表达,抑制经典焦亡途径,诱导了肿瘤细胞对放疗的耐受性[29]。在非小细胞肺癌中METTL3 以m6A/YTHDF2依赖性转录后修饰方式调控NLRP3 的m6A 水平,从而抑制NLRP3 的表达,抑制NLRP3/caspase1/GSDMD相关经典细胞焦亡信号通路的激活,这一过程赋予了肺癌抗细胞焦亡表型并促进酪氨酸激酶抑制剂耐药[30]。
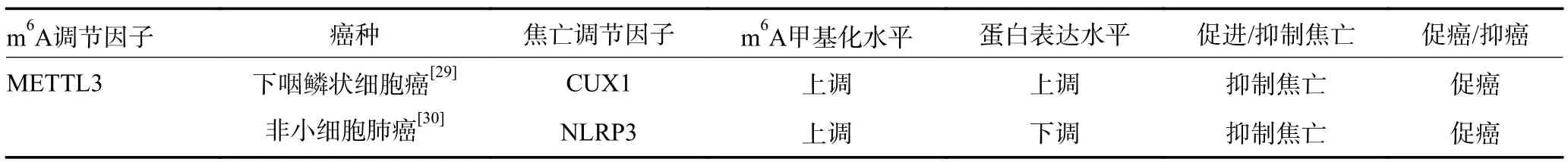
表3 肿瘤细胞焦亡中的m6A 甲基化调控
4 m6A 修饰与肿瘤细胞铁死亡
铁死亡(ferroptosis)是一种铁依赖性程序性细胞死亡调控形式,细胞内铁依赖性的脂质过氧化物的堆积诱导细胞质内的ROS 聚集进而诱导细胞铁死亡。目前已知的铁死亡抑制途径包括细胞内谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)-GSH 系统、铁死亡抑制蛋白1(ferroptosis suppressor protein,FSP1)-CoQ10 系统、二氢乳清酸脱氢酶(dihydroorotate dehydrogenase,DHODH)-CoQH2 系统和GTP 环化酶1(GTP cyclohydrolase 1,GCH1)-四氢生物蝶呤(tetrahydrobiopterin,BH4)系统。研究表明m6A 修饰通过调节细胞内抑制铁死亡的GPX4-GSH 系统、FSP1-CoQ10 系统及其他铁死亡抑制信号活性参与肿瘤细胞铁死亡过程(表4)。
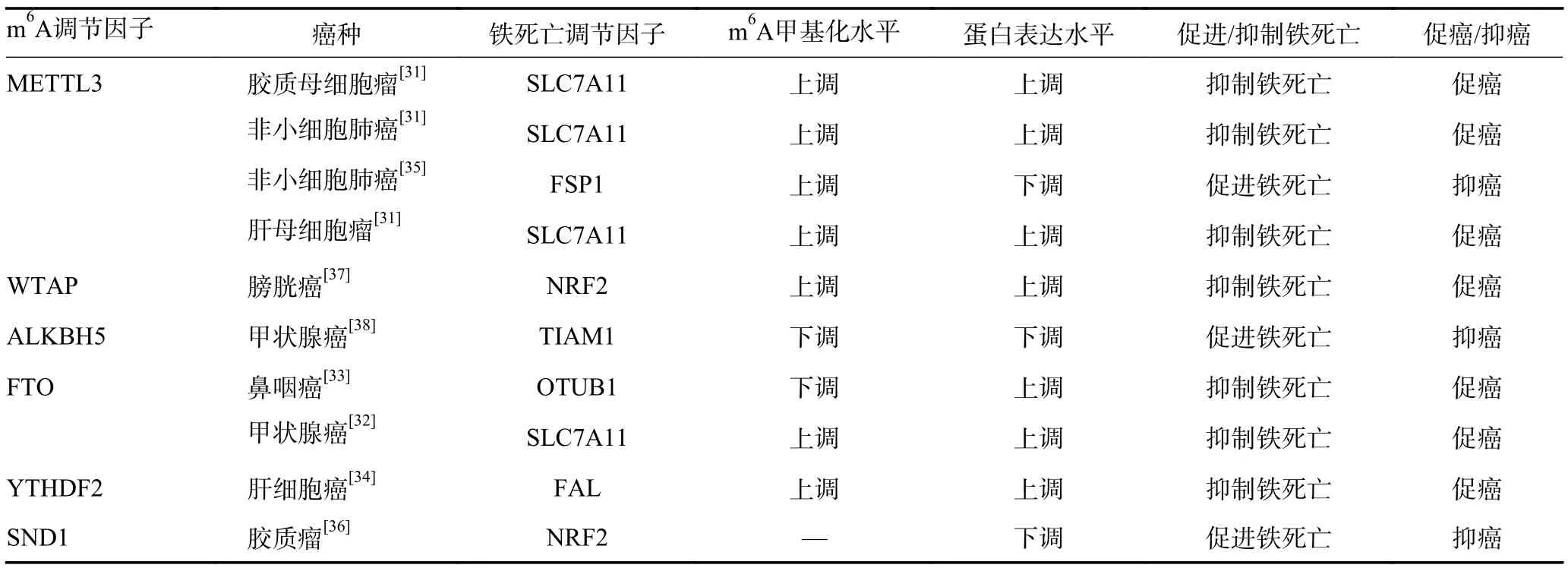
表4 肿瘤细胞铁死亡中的m6A 甲基化调控
4.1 抑制肿瘤细胞铁死亡
4.1.1 调节GPX4-GSH 系统抑制铁死亡 GPX4 和细胞膜上的胱氨酸/谷氨酸转运受体(SystemXc-)通过影响脂质和ROS 代谢抑制铁死亡过程,溶质运载家族7 成员11(solute carrier family 7 member 11,SLC7-A11)作为谷胱甘肽生物合成的胱氨酸转运体通过GPX4-GSH 系统参与铁死亡抑制。在肺腺癌、肝母细胞瘤及胶质母细胞瘤中SLC7A11 是METTL3 的直接靶标,METTL3 介导的m6A 修饰可稳定SLC7A11 mRNA 并促进其翻译,进而抑制肿瘤细胞铁死亡并促进肿瘤细胞增殖,促进肿瘤进展[31]。在甲状腺癌中FTO 介导SLC7A11 的去甲基化,肿瘤细胞中低表达的FTO 增强了SLC7A11 mRNA 的m6A 修饰,刺激SLC7A11 的表观遗传激活,进而抑制肿瘤细胞铁死亡,在甲状腺癌中上调FTO 可促进甲状腺癌中的铁死亡并抑制甲状腺癌的生长[32]。此外,FTO 介导鼻咽癌中OTUB1 mRNA 的m6A 去甲基化,促进OTUB1 表达,OTUB1 通过稳定SLC7A11 抑制辐射诱导的细胞铁死亡,诱发鼻咽癌的辐射抗性[33]。
4.1.2 调节FSP1-CoQ10 系统抑制铁死亡 铁死亡抑制蛋白(ferroptosis suppressor protein 1,FSP1)具有烟酰胺腺嘌呤二核苷酸-泛醌氧化还原活性,不仅能够催化泛醌还原为泛醇来抑制脂质过氧化,还能恢复维生素E 的抗氧化活性而终止脂质过氧化反应,抑制铁死亡。YTHDF2 以m6A 依赖性方式增加了肝细胞癌中铁死亡相关长链非编码RNA FAL 的剪接,上调肝细胞癌中FAL 表达,FAL 通过直接与FSP1 结合竞争性地消除三结构域家族蛋白TRIM69 依赖性FSP1 多泛素化降解来降低铁死亡的脆弱性[34]。非小细胞肺癌组织来源的外泌体miR-4 443 通过靶向抑制METTL3降低顺铂耐药肿瘤细胞中FSP1 的m6A 甲基化水平,在mRNA 和蛋白质水平上增强FSP1 的表达,抑制顺铂诱导的铁死亡,诱导非小细胞肺癌顺铂耐药[35]。
4.1.3 调节其他铁死亡抑制信号 核因子E2 相关因子2(nuclear factor erythroid 2-related factor 2,NRF2)控制并调节谷胱甘肽合成酶、SLC7A11、GPX4、谷氨酸/半胱氨酸连接酶催化亚基以及谷氨酸/半胱氨酸连接酶调节亚基的表达,是脂质过氧化的关键抑制因子,NRF2 mRNA 的3’UTR 发生的m6A 修饰与mRNA的稳定性密切相关。胶质瘤中长链非编码RNA SNAI3-AS1 干扰m6A 阅读蛋白葡萄球菌核酸酶样结构蛋白1(staphylococcal nuclease domain-containing 1,SND1)对NRF2 转录本3’UTR 的m6A 识别,降低NRF2 的mRNA 稳定性,从而促进肿瘤细胞的铁死亡[36]。膀胱癌中WTAP 介导NRF2 mRNA 的m6A 甲基化修饰,经YTHDF1 识别后增强转录本稳定性,抑制肿瘤细胞铁死亡[37]。在甲状腺癌中过表达ALKBH5 可通过降低T 淋巴瘤侵袭转移诱导基因1(T lymphoma invasion and metastasis inducing factor 1,TIAM1)转录本的m6A 水平抑制TIAM1 的表达,进而抑制TIAM1通过调控下游NRF2/血红素加氧酶1 轴诱导的肿瘤细胞铁死亡过程[38]。
5 m6A 修饰与肿瘤细胞坏死性凋亡
坏死性凋亡,又称细胞程序性坏死,是一种受调控的坏死类型,其信号级联导致形成二硫键依赖性的混合系激酶结构域样蛋白淀粉样聚合物,从而导致炎性细胞膜的破坏,激活炎性细胞死亡机制。m6A 修饰可调节机体细胞坏死性凋亡过程,研究发现核不均一核糖核蛋白C(heterogeneous nuclear ribonucleoprotein C,HNRNPC)促进ATF4 mRNA 的m6A 修饰,刺激甲状腺滤泡上皮细胞的坏死性凋亡,进而介导自身免疫性甲状腺疾病的发生[39]。在腹主动脉瘤中,METTL3/METTL14 复合物介导受体相互作用蛋白3(receptor interacting protein 3,RIP3)mRNA 的m6A修饰,经m6A 修饰的RIP3 转录本与YTHDF3 结合后增加RIP3 蛋白表达水平,诱导血管平滑肌细胞坏死性凋亡[40]。目前研究证实坏死性凋亡信号在促进肿瘤发展、肿瘤转移等方面发挥重要作用且肿瘤细胞坏死性凋亡的激活可增强机体抗肿瘤免疫,m6A 修饰同样参与肿瘤细胞坏死性凋亡过程。Lan 等[41]发现METTL3通过m6A 依赖性方式抑制执行坏死性凋亡的关键亚基肿瘤坏死因子受体相关因子5(tumor necrosis factor receptor-associated factor 5,TRAF5)介导体内外结肠癌细胞的坏死性凋亡,进而诱导结直肠癌细胞对奥沙利铂的耐药性。
6 m6A 修饰与肿瘤细胞铜死亡
铜死亡是一种新发现的由过量铜引发的程序性细胞死亡形式,该过程是由线粒体中Cu2+与脂酰化蛋白靶向结合,导致靶蛋白聚集,干扰线粒体呼吸链的正常运行,最终诱导细胞死亡。铜死亡参与了肿瘤发生和转移的大部分机制,并调节肿瘤免疫逃逸。铁氧还蛋白1(ferredoxin 1,FDX1)是蛋白脂酰化的上游调节因子,可以促进二氢脂酰胺S-乙酰转移酶和二氢硫代酰胺S-琥珀酰基转移酶的脂质酰化,是线粒体呼吸链的关键因素,FDX1 基因敲除导致蛋白质脂酰化完全丧失,细胞铜死亡抗性明显增强。研究发现,C-MYC 可以靶向与YTHDF1 结合,促进YTHDF1对FDX1 转录本m6A 甲基化的识别,上调FDX1 表达,这与胶质瘤细胞对铜死亡的敏感性高度相关。铜死亡相关基因SLC31A1 同样参与铜死亡过程,目前被认为是预测乳腺癌诊断、预后的治疗反应的潜在指标。Lian 等[42]使用数据库分析发现SLC31A1 的表达与YTHDF3、YTHDF2、RBM15 和YTHDF1 呈强正相关,YTHDF3 可能通过m6A 依赖性方式上调SLC-31A1 表达进而促进乳腺癌脑转移,但该过程仍待进一步研究证实。
7 结语与展望
随着各类肿瘤发病率的上升,肿瘤治疗一直是临床发展的重点。随着肿瘤基因谱的完善,肿瘤治疗已从放化疗发展到靶向治疗、免疫治疗和其他个性化的治疗方法。早期肿瘤的治疗方法主要是阻止肿瘤细胞的生物合成,降低其繁殖和转移能力。随着研究的深入,人们逐渐意识到促进肿瘤细胞死亡也是治疗肿瘤的可行的方法,并且与不受机体调控的意外细胞死亡相比,程序性细胞死亡可以通过药物干预调节。目前已有诱导程序性细胞死亡的表观遗传调节剂应用于临床。Vorinostat(伏立诺他)是一种广谱组蛋白去乙酰化酶抑制剂,可抑制关键自噬标志物的去乙酰化,从而干扰肿瘤细胞自噬;Panobinostat(帕比司他)抑制组蛋白乙酰化酶,重建肿瘤细胞中的正常基因表达并有助于驱动多种信号通路,包括诱导组蛋白乙酰化,促进caspase3/7 活性、降低抗凋亡因子(Bcl-2、Bcl-XL)水平等促进细胞凋亡的外在和内在途径。作为真核生物体内最丰富的转录后表观遗传调控方式,m6A 修饰在肿瘤细胞凋亡、自噬、焦亡、铁死亡、坏死性凋亡、铜死亡中发挥重要作用,通过调节m6A 修饰水平诱导肿瘤细胞死亡是潜在的肿瘤治疗策略。
根据程序性细胞死亡分子机制,除凋亡、自噬、焦亡、铁死亡、坏死性凋亡、铜死亡外还有内源性细胞死亡、网状细胞死亡、PARP-1 依赖性细胞死亡、溶酶体依赖性细胞死亡、细胞内碱化死亡、氧自由基诱导死亡等程序性细胞死亡方式。这些程序性细胞死亡方式同样受表观遗传调控,例如:赖氨酸去甲基化酶6B介导DNA 损伤诱导的PARP-1 依赖性细胞死亡;组蛋白H3 赖氨酸27 特异性甲基转移酶Zeste 增强子同源物2 介导巨噬细胞诱导的间皮瘤细胞氧自由基诱导死亡,但其与m6A 修饰的相互联系仍待挖掘。进一步探索m6A 修饰在诱导程序性细胞死亡中的作用机制,构建调控网络,开发针对异常m6A 修饰的表观遗传调节剂,进而精准调控肿瘤程序性细胞死亡途径介导细胞死亡是肿瘤治疗的潜在研究方向。
本文无影响其科学性与可信度的经济利益冲突。
