基于加权基因共表达网络探索喉癌干性关键基因
2024-04-08昝雨欣
昝雨欣,丁 妍
1. 湖北医药学院生物医药研究院(湖北十堰 442000) 2. 湖北医药学院附属太和医院生命科学研究所(湖北十堰 442000) 3. 胚胎干细胞研究湖北省重点实验室(湖北十堰 442000)
喉癌是耳鼻喉科最常见的恶性肿瘤,好发于欧洲地区[1]。目前喉癌的治疗手段包括手术、放化疗及生物治疗等,虽然有一定疗效,但喉癌患者的5年生存率仅为30%~40%,且大多数患者在确诊时往往处于晚期,多死于肿瘤复发或转移[2]。因此,亟待找到一种有效可靠的方法对喉癌患者进行早期诊治,以改善预后。
肿瘤干细胞是一类具有自我更新和分化能力的异质性肿瘤细胞,在肿瘤的增殖、转移和复发中发挥重要作用[3]。通过人工智能和深度学习算法,建立一种描述肿瘤干细胞的新方法,称为肿瘤干性指数,用于描述肿瘤细胞和干细胞之间的相似程度。研究表明,mRNAsi 可能是肿瘤患者生存、分类和疾病进展的有效指标[4]。目前对喉癌的认识主要依赖于头颈部相关研究,其独立研究相对缺乏,且基于生物信息学探索喉癌与mRNAsi 潜在联系的研究较少。本研究采用加权基因共表达网络分析(weighted gene co-expression network analysis, WGCNA),基于大规模数据集识别喉癌潜在相关基因模块,旨在识别mRNAsi 基因潜在生物标志物或治疗靶点,并验证与喉癌免疫预后相关的新生物标志物[5]。
1 资料与方法
1.1 数据来源和处理
从癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库(https://www.cancer.gov/ccg/research/genome-sequencing/tcga)中获取11 个正常样本和112 个喉癌样本的FPKM 数据类型和相应临床特征的RNA 测序数据。采用R 软件的limma 软件包筛选喉癌和正常组织之间的差异表达基因,错误发现率(false discovery rate, FDR)<0.05 且|log(FC)|>1 被认为是显著的。
1.2 WGCNA网络构建
WGCNA 是一种评估基因之间相关性并将高度相关基因分类到同一模块的方法。首先通过差异表达分析进行处理,以过滤无关信息。拓扑重叠测度用于识别共表达基因模块,以降低网络对虚假连接的敏感性。通过将聚类树切割成分支,将具有高度绝对相关性的基因聚类到相同模块。只有超过50 个基因才会被定义为一个模块。具有较高相关性的模块将被合并(r<0.25),每个模块被分配不同颜色进行可视化。
1.3 关键模块和基因筛选
为了评估模块的显著性,将基因与样本性状之间的相关性计算为基因显著性(gene significance,GS)。模块自身基因和基因表达谱之间的相关性被定义为模块成员(module membership, MM)。选择mRNAsi 和表观遗传调控的mRNAsi(EGERmRNAsi)作为临床表型进一步分析。|r|>0.3 被认为具有相关性。
1.4 风险模型构建
首先进行单因素Cox 回归分析,以评估预后相关模块的mRNA,再采用LASSO 回归分析确定mRNA 系数,采用以下算法计算患者的风险得分:
风险评分=∑ni=coefi×mRNA 表达
采用Rtsne 和ggplot2 R 软件包,在mRNAs 特征表达的前提下进行主成分分析(principal component analysis, PCA),以区分喉癌患者的高低风险队列。
1.5 药物敏感性分析
在CellMiner 数据库(https://discover.nci.nih.gov/cellminer/home.do)[6]下载常用药物敏感性和基因表达数据,与上述风险系数进行相关性分析,通过R 语言中impute、limma、ggpubr、ggplot2 包筛选具有高评分的基因和对应的药物。
1.6 免疫相关性分析
采用R 包GSVA 对免疫细胞和功能的基因表达谱进行单样本基因集富集分析(single sample gene set enrichment analysis,ssGSEA),采用ggpubr 包将结果可视化。同时通过肿瘤免疫功能障碍与排斥(Tumor Immune Dysfunction and Exclusion, TIDE)数据库(http://tide.dfci.harvard.edu)下载肿瘤评分数据,采用limma、ggpubr 软件包得到高低风险基因与肿瘤免疫逃逸结果。
1.7 功能注释
利用clusterProfiler 包对高低风险表达组进行基因集富集分析(gene set enrichment analysis,GSEA),下载MSigDB 数据库中的“c2.cp.kegg.v7.5.1.entrez.gmt”和“c5.go.v7.5.1.symbols.gmt”基因集,进行基因本体(gene ontology, GO)分析及京都基因和基因组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)通路富集分析,将FDR <0.05 设置为阈值。
1.8 统计分析
采用R 4.0.3 软件进行统计分析。分类变量采用χ2检验,连续变量采用秩检验。采用Pearson相关性分析评估关键基因的表达与药物敏感性之间的相关性。以P<0.05 为差异具有统计学意义。
2 结果
2.1 mRNAsi相关差异表达基因
为进一步探讨与mRNAsi 相关的功能基因,计算正常组织和喉癌组织之间存在的显著差异,红色为高表达基因,绿色为低表达基因。共筛选得到5 302 个差异表达基因,并绘制火山图,见图1-A。其中,前14 个差异表达基因热图见图1-B,KRT4、CRNIN、MAL等基因在正常组织中高表达(P<0.05)。
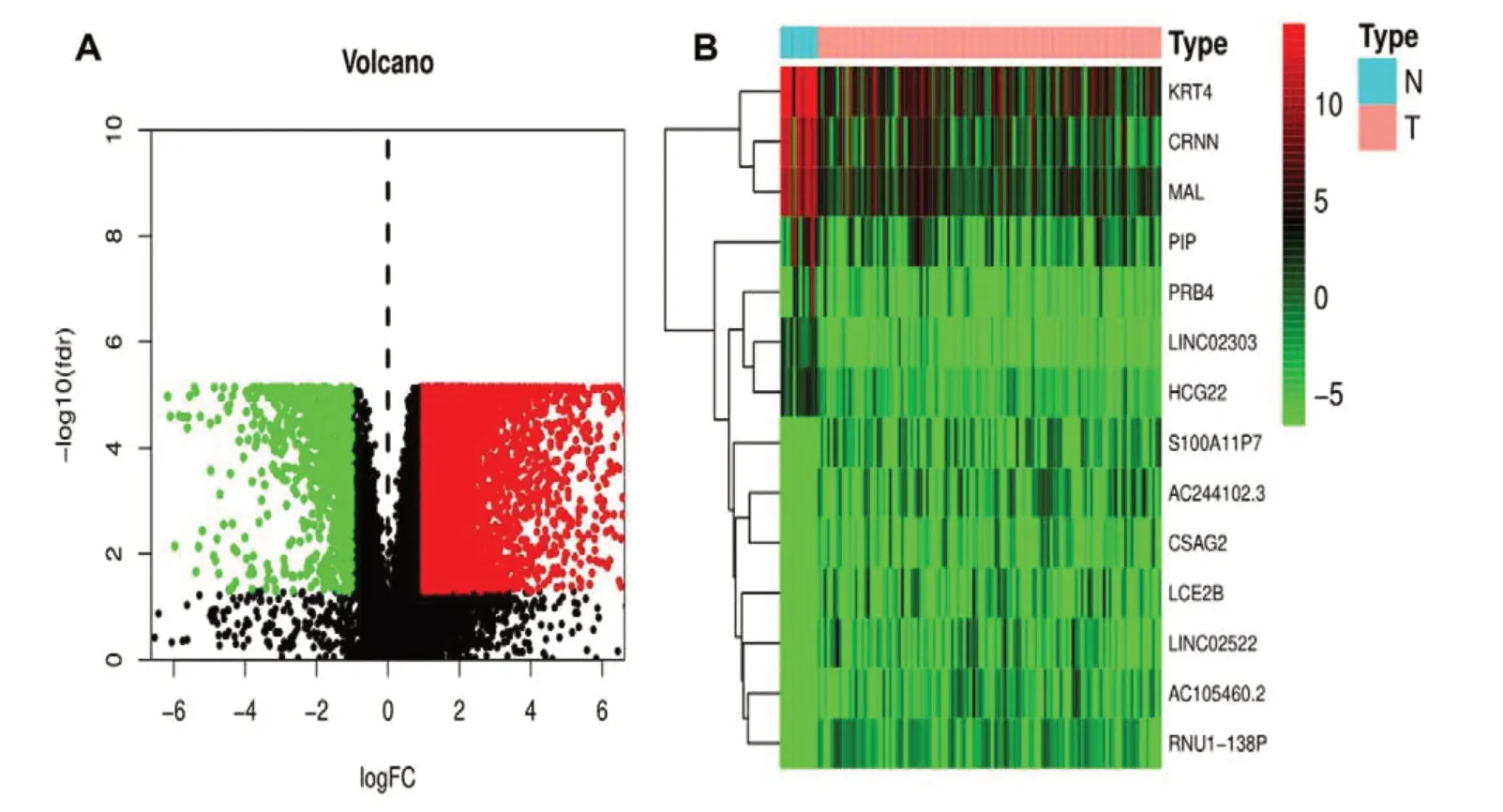
图1 喉癌的差异表达基因Figure 1. Differentially expressed genes in laryngeal cancer注:A. 火山图;B. 前14个差异表达基因热图。
2.2 WGCNA筛选的重要模块与基因
选择上述差异表达基因作为WGCNA 分析的输入端数据,构建一个基因共表达网络并从中识别与mRNAsi / EREG-mRNAsi 显著关联的基因模块,软阈值(β=5,R2=0.9)用于保证无规模网络,最终获得30 个符合要求的模块。然后,对相关模块的全基因表达水平进行分析,以确定相应模块与mRNAsi 之间的相关性,见图2-A。在30 个模块中,紫色模块与喉癌样本的mRNAsi分值呈显著正相关(MS=0.53,P<0.05),同时随机选择另一种正相关的模块(浅黄色模块)加以验证。紫色模块内共有7 个显著差异表达基因(GS >0.5 且MM >0.7),见图2-B。浅黄色模块内共有4 个显著差异表达基因(GS >0.3 且MM >0.8),见图2-C。最后,选择上述浅黄色模块差异表达基因(MTHFD2、LINC01932、AL121761.1、TBX2)和紫色模块差异表达基因(NEK2、KPNA2、ASF1B、MCM10、RFC5、FAM72A、DBF4)共11个基因作为喉癌的关键基因。
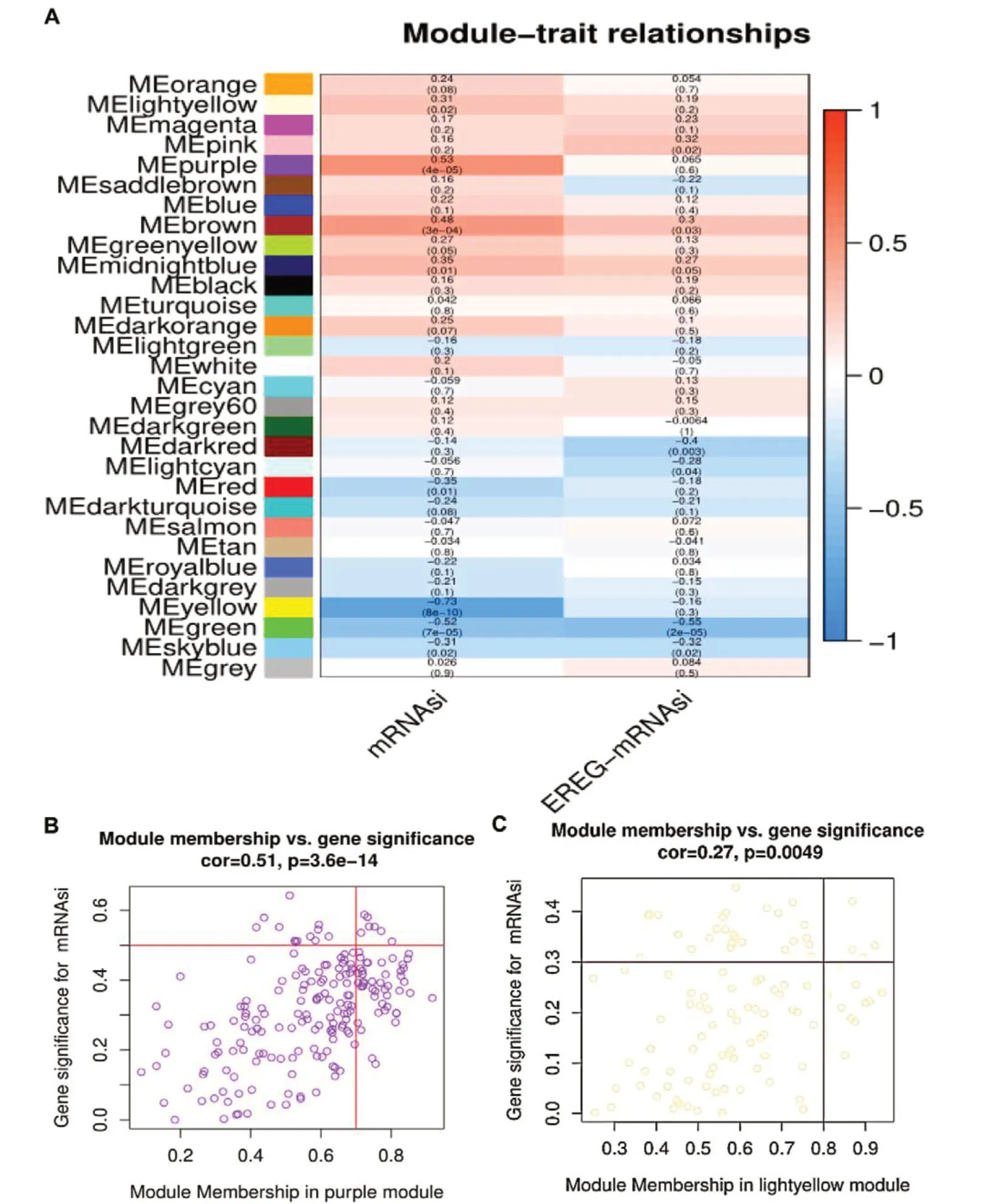
图2 WGCNA筛选的重要模块与基因Figure 2. The important modules and genes for WGCNA screening注:A. 模块-mRNAsi(EREG-mRNAsi)关联图;B. 紫色模块散点图;C. 浅黄色模块散点图。
2.3 预后基因筛选与验证
进一步绘制11 个基因在正常组织和肿瘤组织中的表达趋势图,发现浅黄色、紫色模块中的候选基因在喉癌中大多上调(P<0.05),见图3-A。模块内基因相关性分析结果显示,紫色模块协同表达关系较强的基因对共有两对,包括FAM72A和NEK2(r=0.77,P<0.05)、MCM10和RFC5(r=0.71,P<0.05)。浅黄色模块协同表达关系较强的基因对共有三对,包括MTHFD2和TBX2(r=0.74,P<0.05)、LINC01932和TBX2(r=0.71,P<0.05)、LINC01932和MTHFD2(r=0.70,P<0.05),见图3-B。对11 个基因进行Cox 回归和LASSO 回归分析,构建预后基因风险模型:风险分数=TBX2×0.021+MTHFD2×0.085,见图3-C。PCA 分析结果表明,所得的风险评分有效区分了高低风险组,见图3-D。通过化疗药物数据库筛选出前12 个结果进行可视化,结果显示TBX2基因与维莫非尼(Vemurafenib)(r=0.587,P<0.001)、达拉菲尼(Dabrafenib)(r=0.439,P<0.001)均呈正相关;MTHFD2基因与奥沙利铂(Oxaliplatin)(r=0.368,P=0.004)呈正相关,与凡德他尼(Vandetanib)(r=-0.354,P=0.005)呈负相关,见图3-E。

图3 喉癌与mRNAsi相关的关键基因表达Figure 3. Expression of key genes related to mRNAsi in laryngeal cancer注:A. 模块基因表达水平;B. 模块基因的相关性;C. 单变量Cox回归分析;D. 基于风险评分的PCA图和t-SNE图;E. 化疗药物筛选;*P<0.05,***P<0.001。
2.4 预后基因免疫微环境
探索风险评分与mRNAsi 的相关性,其与RNAss 呈正相关(r=0.35,P<0.05),与DNAss 的相关性差异无统计学意义(r=0.073,P=0.44),见图4-A。同时检测风险评分与肿瘤免疫逃逸的相关性,发现高低风险组存在显著差异。TIDE 是一种模拟肿瘤免疫逃逸机制的计算方法,以预测样本对免疫检查点抑制剂的反应。两基因构建的低风险组TIDE 明显高于高风险组(P<0.001),见图4-B。在低风险队列中,浆细胞样树突状细胞(plasmacytoid dendritic cells,pDCs)和辅助性T 细胞2(T helper 2 cell,Th2)的比例增加,见图4-C。在低风险队列中,趋化因子受体(chemokine receptor,CCR)、人类白细胞抗原(human leukocyte antigen,HLA)及T 细胞共刺激(T cell co-stimulation) 3 种免疫功能均有差异表达,见图4-D。
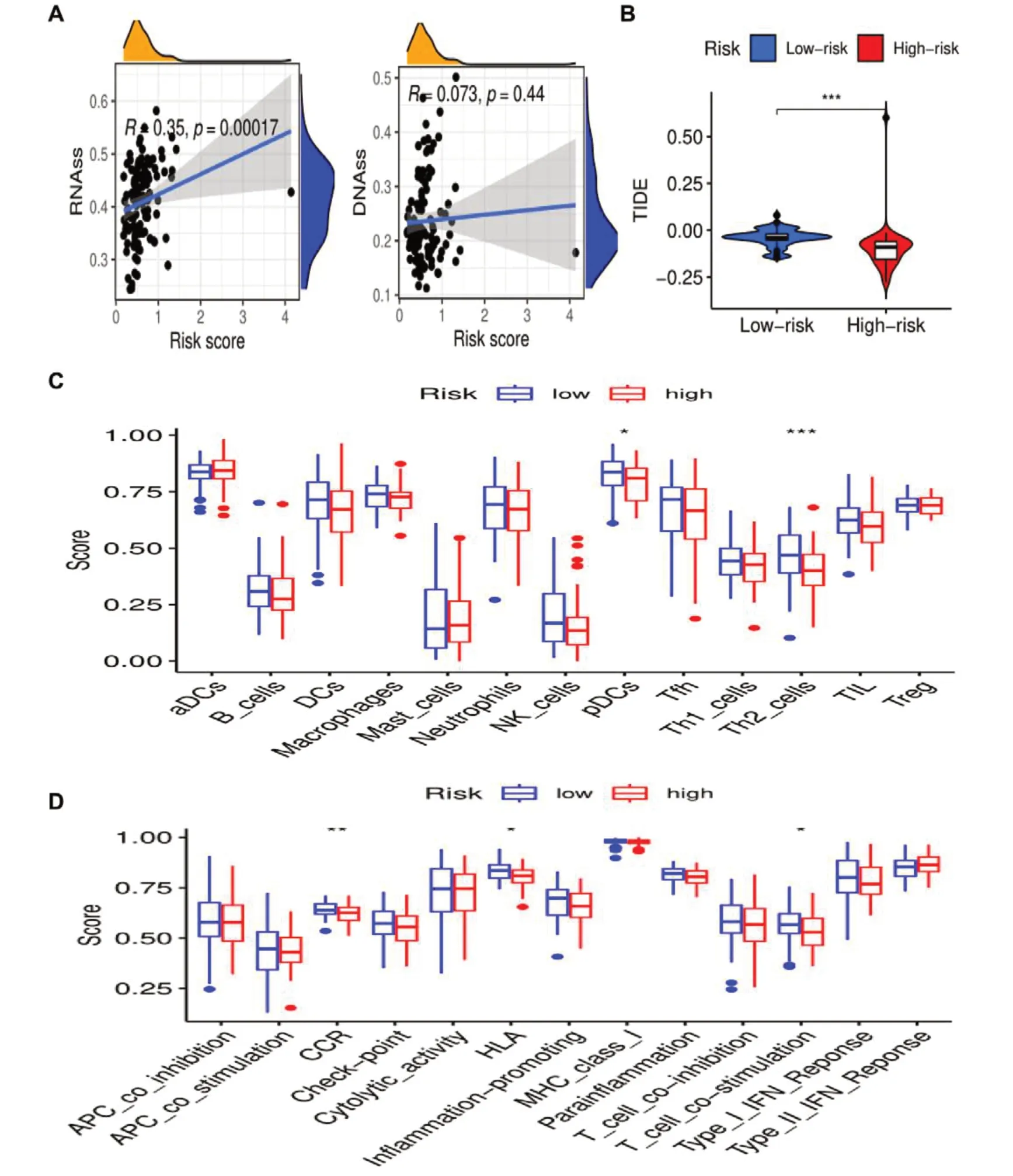
图4 喉癌特异性肿瘤微环境Figure 4. Laryngeal carcinoma specific tumor microenvironment注:A. 风险曲线与mRNAsi相关性;B. 风险评分与TIDE的关系;C、D. 低风险和高风险之间免疫相关细胞与途径的ssGSEA富集分数比较队列;*P<0.05,**P<0.01,***P<0.001。
2.5 GO和KEGG富集分析
GO 功能富集分析结果显示,MTHFD2主要富集于表皮细胞分化,TBX2主要富集于结缔组织替代的正向调节,且二者与角化作用、角化细胞分化密切相关,见图5-A。KEGG 信号通路富集分析结果显示,MTHFD2主要富集于嗅觉信号转导途径,TBX2主要富集于同种异体移植排斥反应和蛋白酶体,见图5-B。
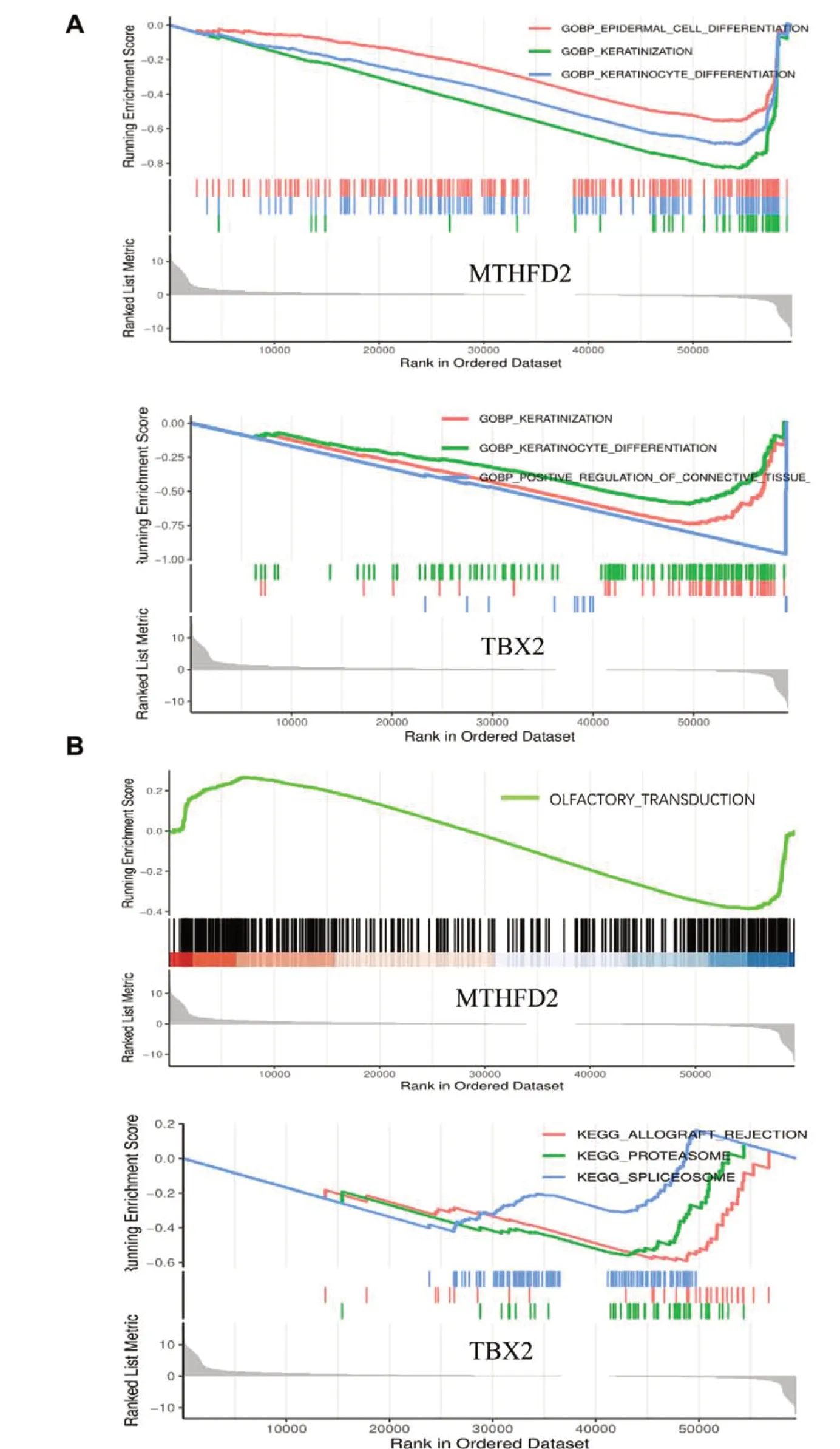
图5 MTHFD2与TBX2基因功能富集分析Figure 5. Functional enrichment analysis of MTHFD2 and TBX2 genes注:A. GO富集分析;B. KEGG富集分析。
3 讨论
喉癌的发病率较低,每10 万人中仅有1.5 例发病,临床较少见[1]。随着人口老龄化,喉癌的发病率逐渐上升,喉癌患者在生物学行为、治疗反应和预后方面的表现具有较大异质性。年龄、肿瘤分级和TNM 分类等常见的预后因素无法准确预测喉癌患者的预后,因而聚焦该疾病具有重要的临床实际意义。
肿瘤干细胞是恶性肿瘤无限增殖和复发的来源。然而,早期研究缺乏对癌症干细胞的统一描述,喉癌的个体化治疗进展缓慢。而mRNAsi 作为量化肿瘤患者肿瘤干性的指标被开发出来,可以更加高效便捷地探索与肿瘤干性相关的基因。肿瘤干细胞在喉癌中的研究仍处于初级阶段。Prince 等曾报道在原发性喉癌中检测到CD44+癌细胞,虽然其占比不到10%,但在体外具有很强的成瘤能力[7]。研究表明,CD133 是喉癌干细胞的标志物之一,可以为靶向治疗提供证据[8]。这表明肿瘤干细胞标记物与喉癌治疗相结合,可能对喉癌患者的长期预后产生积极影响,并为癌症治疗带来新思路。本研究构建了一个基于WGCNA 的mRNAsi 的喉癌预测模型,得到了关键基因MTHFD2和TBX2,为喉癌的机制研究提供了新见解。
mRNAsi 相关基因的药敏数据将有助于喉癌患者的治疗。与MTHFD2相关的前两种化疗药物维莫非尼和达拉菲尼均为有效的丝氨酸/苏氨酸蛋白激酶选择性抑制剂,且前者在甲状腺乳头状癌患者中显示出抗肿瘤活性,后者可抑制成釉细胞癌与甲状腺癌进展[9-10]。奥沙利铂和凡德他尼是两种与TBX2相关的化疗药物,奥沙利铂是一种DNA 合成抑制剂,可以诱导免疫原细胞死亡,并对喉癌细胞具有抑制作用[11];凡德他尼是一种多通道肿瘤信号传导抑制剂,通过调节细胞依赖性DNA 修复和肿瘤微环境,使头颈鳞癌对光动力学治疗过敏,从而导致体内肿瘤生长的显著增加[12]。双基因共相关的达沙替尼是一种具有抗实体肿瘤双重SRC/ABL 激酶抑制剂,抑制SRC 磷酸化反应诱导细胞周期停止和凋亡,从而抑制肿瘤进展[13]。这些药物是喉癌患者的潜在新治疗选择。
肿瘤微环境的免疫细胞数量和比例随着肿瘤进展而变化,在某些情况下可以对抗癌治疗产生反应。丰富的抗肿瘤浸润免疫细胞(pDCs 和Th2)富集在低风险队列中,pDCs 和Th2 细胞均是参与免疫反应并在抗肿瘤反应中发挥积极作用的主要细胞。在扁桃体上皮中,pDCs 不仅可以通过识别病毒并释放干扰素帮助抵御病毒感染,而且能够在不受微生物过度刺激的情况下启动免疫。因此,pDCs可以被认为是扁桃体上皮的“守护者”,帮助维护免疫系统的稳定和健康[14]。此外,临床证据显示,使用pDCs 的药物分类法识别恶性肿瘤治疗敏感的患者,可有效提高治疗反应率[15]。以上均提示pDCs 在喉癌进展中发挥抑制作用。在喉鳞状细胞癌患者中,Th2 型转录因子和细胞因子的表达占优势[16]。经批准的免疫检查点抑制剂可减少免疫逃逸,从而改善癌症患者的临床预后。临床研究发现一例特殊的转移性喉癌病例,患者在PD-1 抑制剂治疗失败后,出现了纵隔腹膜后淋巴结和骨质进行性疾病,但在经过免疫检查点抑制剂药物治疗后,其骨痛、腹膜后淋巴结和胸腔淋巴结病症状得到缓解,临床状况也得到改善,这与TIDE 评分结果一致[17],有助于开发个性化和精确的免疫治疗方案。在功能富集研究中,发现关键基因与角化细胞密切相关,而喉癌前病变喉角化症是一种常见的粘膜病变,起源于造血干细胞的细胞缺陷会导致喉角化状癌发生,这提示未来治疗可通过免疫学对恶性肿瘤进行早期监测[18]。
本研究存在一定局限性,尽管采用了生物信息学分析,但实际临床研究的样本量较少,降低了研究的可行性。综上所述,TBX2和MTHFD2基因可成为喉癌早期检测、诊断和预后评估的潜在生物标志物。将WGCNA模块与mRNAsi相结合,通过精准定位这些模块中的关键基因,可以确定在癌症进展中起关键作用的潜在治疗靶点,这些特定基因或途径的靶向可能为开发喉癌新的治疗策略和确定药物靶点提供参考。
