Activation of peroxymonosulfate by FeVO3-x for the degradation of carbamazepine: Vanadium mediated electron shuttle and oxygen vacancy modulated interface chemistry
2024-04-06LeiduoLaiHongyuZhouYihenHongMengfanLuoYangShiHengZhangZhaokunXiongGangYaoBoLai
Leiduo Lai ,Hongyu Zhou ,Yihen Hong ,Mengfan Luo ,Yang Shi,∗ ,Heng Zhang ,Zhaokun Xiong,Gang Yao,Bo Lai,∗
a State Key Laboratory of Hydraulics and Mountain River Engineering,College of Architecture and Environment,Sichuan University,Chengdu 610065,China
b Sino-German Centre for Water and Health Research,Sichuan University,Chengdu 610065,China
c Chengdu Baixi Environmental Technology Company,Chengdu 610065,China
d Institute of Environmental Engineering,RWTH Aachen University,Germany
Keywords: Peroxymonosulfate Fe(III)/Fe(II) cycle Electron shuttles Oxygen vacancy Bimetallic catalysts
ABSTRACT Fast Fe(III)/Fe(II) circulation in heterogeneous peroxymonosulfate (PMS) activation remains as a bottleneck issue that restricts the development of PMS based advanced oxidation processes.Herein,we proposed a facile ammonia reduction strategy and synthesized a novel FeVO3-x catalysts to activate PMS for the degradation of a typical pharmaceutical,carbamazepine (CBZ).Rapid CBZ removal could be achieved within 10 min,which outperforms most of the other iron or vanadium-based catalysts.Electron paramagnetic resonance analysis and chemical probe experiments revealed SO4•-,•OH,O2•- and high valent iron (Fe(IV)) were all generated in this system,but SO4•- and Fe(IV) primarily contributed to the degradation of CBZ.Besides,X-ray photoelectron spectroscopy and X-ray adsorption spectroscopy indicated that both the generated low-valent V provides and oxygen vacancy acted as superior electron donors and accelerated internal electron transfer via the unsaturated V-O-Fe bond.Finally,the proposed system also exhibited satisfactory performance in practical applications.This work provides a promising platform in heterogeneous PMS activation.
Iron induced heterogeneous peroxymonosulfate (PMS) activation processes are promising technologies for the degradation of personal care products (PPCPs) in waters due to the strong oxidation capacity of the generated reactive oxygen species (ROS) [1–3].However,the rate-limiting step of Fe(III) conversion to Fe(II) significantly prohibits PMS decomposition and ROS generation,thus inhibiting the degradation of target pollutants [4,5].In order to expedite the Fe(III)/Fe(II) cycle and ROS generation,previous works usually introduced homogeneous reducing agents in PMS activation processes as co-catalysts for fast removal of refractory pollutants [6–9].Unfortunately,the introduction of homogeneous reductants is restricted in the practical applications due to the serious environmental pollution induced by the oxidized by-products in waters [10].Therefore,it is of great interest to develop novel strategies to accelerate the Fe(III)/Fe(II) circulation.
It was reported that transition metals serving as electron shuttles can also facilitate the Fe(III)/Fe(II) conversion in Fenton-like oxidation,since the polyvalent metals as electron-sacrificers can donate electrons to Fe(III) until they are oxidized to the highest valence [4,11-13].Our previous results suggested that when the MnFe2O4,Fe2Mo3O12and Fe2TiO5served as catalysts,pollutant degradation efficiencies in PMS activation processes are much lower than that of FeVO4[4],indicating V species serving as electron shuttles outperform Mo,Ti,Mn.Therefore,modulating Febased materials with V species provides a great platform to accelerate Fe(III)/Fe(II) circulation without environmental pollution caused by dissolved matters in waters.Nevertheless,in most cases,stable state of V species in materials are quinquevalent (V(V)) due to the electron-deficient of 3d34s2orbits,implying the difficulties of V species as electron donators lie in the strategy for V reduction in the V modulated Fe-based materials.Our previous study suggested that adding reducing agents can facilitate V(V)species reduction in the FeVO4,further triggering the long-lasting Fe(III)/Fe(II) circulation to effectively degrade target pollutants [4].However,reducing agents at inappropriate dosage may impede the pollutant degradation efficiency due to the quenching effect of ROS and the direct consumption of oxidants.Therefore,it is imperative to develop new tactics to reduce V species in the Fe-V bimetallic materials.
In this work,a low valent Fe-V bimetallic material (FeVO3-x)with enriched oxygen vacancy was prepared by a coprecipitation method and an ammonia reduction method to activate PMS for carbamazepine (CBZ) degradation.Notably,the reduction process would not only lead to the reduction of V,but also induce the generation of abundant electron-rich oxygen vacancy,which was reported to be a robust PMS adsorption site as well as a flexible switcher to regulate the mechanism transformation from radical to high valent iron (Fe(IV)) generation [14–16].Therefore,the FeVO3-xmay exhibit dual functions to activate PMS and generate a myriad of ROS for CBZ degradation: (i) low valent V species as electron donators raise the efficiency of Fe(III)/Fe(II) conversion;(ii) oxygen vacancy facilitates the generation of high valent Fe species.This work provides new insights into the heterogeneous Fe(III) reduction and ROS production by the defect Fe-V bimetallic catalyst.In addition,the CBZ transformation products and their corresponding toxicity were examined to evaluate the practicability of the FeVO3-x/PMS system.
Details about the chemicals were provided in Text S1 (Supporting information).The FeVO3-xwas synthesized by the combination methods of coprecipitation and ammonia reduction.In a typical procedure,2.8078 g NH4VO3was dissolved in 150 mL deionized water at 80 °C,and then 150 mL of transparent orange solution with 9.696 g Fe(NO3)3·9H2O was slowly added into NH4VO3solution and stirred for 1 h at 80 °C.The solution was further modulated with dilute ammonia to maintain the pH at 8.0,and aged for several hours at room temperature.Then,the precipitates after filtration and washing with ethanol and water were dried at 110 °C and then calcined in the air at 600 °C for 4 h.Finally,the obtained precipitates were further calcined under ammonia atmosphere with nitrogen purging (10%,N2as the carrier gas,the tube furnace is connected with a washing bottle filled with ammonia)at 700 °C for 4 h to constitute a defect low valent Fe-V bimetallic catalyst.
For pollutant degradation,all bath experiments were conducted with 150 mL target pollutant solution.The reaction was initiated by adding predetermined amounts of catalysts and PMS.The solution temperature was adjusted at 30 ± 1 °C by using water batch.At the predetermined time,certain volume of reaction solution was filteredviaa 0.22 μm PTFE syringe filter discs and mixed with 20 μL Na2S2O3before analysis.Details about the characterization and analytic methods could be seen in Text S2 (Supporting information).
X-ray diffraction (XRD) spectrum suggests that the distinct diffraction peaks of FeVO3-xare indexed to (104),(311),(110),and(116) planes,and the crystal phases of pristine FeVO3-xparticles are assigned toγ-Fe2O3and V2O3(Fig.1a),indicating low valent V species exist in FeVO3-x.The vibrating sample magnetometer (VSM) results shows the saturation magnetization of FeVO3-xis ∼3 emu/g (Fig.1b),which might originate from the ferromagnetism ofγ-Fe2O3.As shown in Fig.1c,the fresh FeVO3-xparticles are micron-sized and exhibit a unique brain-like morphology with wrinkled surface,which would facilitate the adhesion of PMS in the vicinity of FeVO3-x.Accordingly,the mappings of Fe,V,O species (Figs.1d-f) show that only Fe and V species are uniformly distributed on FeVO3-x,indicating enriched oxygen vacancy was created on the surface of FeVO3-x.Moreover,since the photoluminescence (PL) emission peak at 450 nm is correlated with the electron transition from shallow level to the top of the valence band,the existence of oxygen vacancy in the FeVO3-xis also verified by PL spectrum (Fig.S1 in Supporting information) [17].In addition,the N2adsorption-desorption results (Fig.S2 in Supporting information) manifest that the surface area of fresh FeVO3-xis 1.1 m2/g,suggesting the poor pollutant adsorption ability of FeVO3-x.

Fig.1.(a) XRD spectrum of FeVO3-x.(b) The magnetic hysteresis loop of FeVO3-x.(c) SEM image and (d-f) the corresponding element mappings of FeVO3-x.
The performance of FeVO3-x/PMS system was compared with several systems,including FeO/PMS,Fe3O4/PMS,Fe2O3/PMS,V2O3/PMS,V2O4/PMS,FeVO3-xalone and PMS alone systems.Results shown in Fig.2a and Fig.S3 (Supporting information) suggest that PMS in the absence of catalyst cannot oxide CBZ,and FeVO3-xhas no adsorption effect on CBZ,indicating CBZ was degraded by the generated ROS in the FeVO3-x/PMS system.In addition,the CBZ degradation efficiency in the FeVO3-x/PMS system within 10 min treatment is much higher (100%) than those in other systems (6%-36%),implying FeVO3-xcan effectively activate PMS to generate substantial ROS for CBZ degradation.
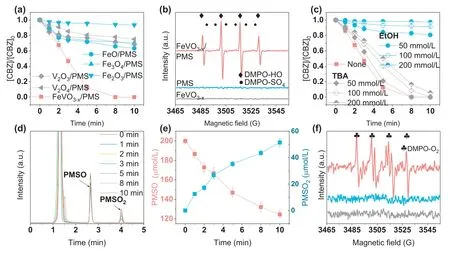
Fig.2.(a) CBZ degradation in different systems.(b) DMPO-HO and DMPO-SO4 adducts in the FeVO3-x/PMS system.(c) Quenching effects of TBA and EtOH on CBZ degradation.(d) HPLC chromatograms of PMSO and PMSO2 in the FeVO3-x/PMS system.(e) PMSO degradation and PMSO2 generation in the FeVO3-x/PMS system.(f) DMPO-O2 adducts in the FeVO3-x/PMS system.Experiment condition: [FeVO3-x]0=100 mg/L,[PMS]0=0.2 mmol/L,[CBZ]0=2.5 mg/L,pHini=6.3,[PMSO]0=200 μmol/L,[DMPO]0=50 mmol/L.
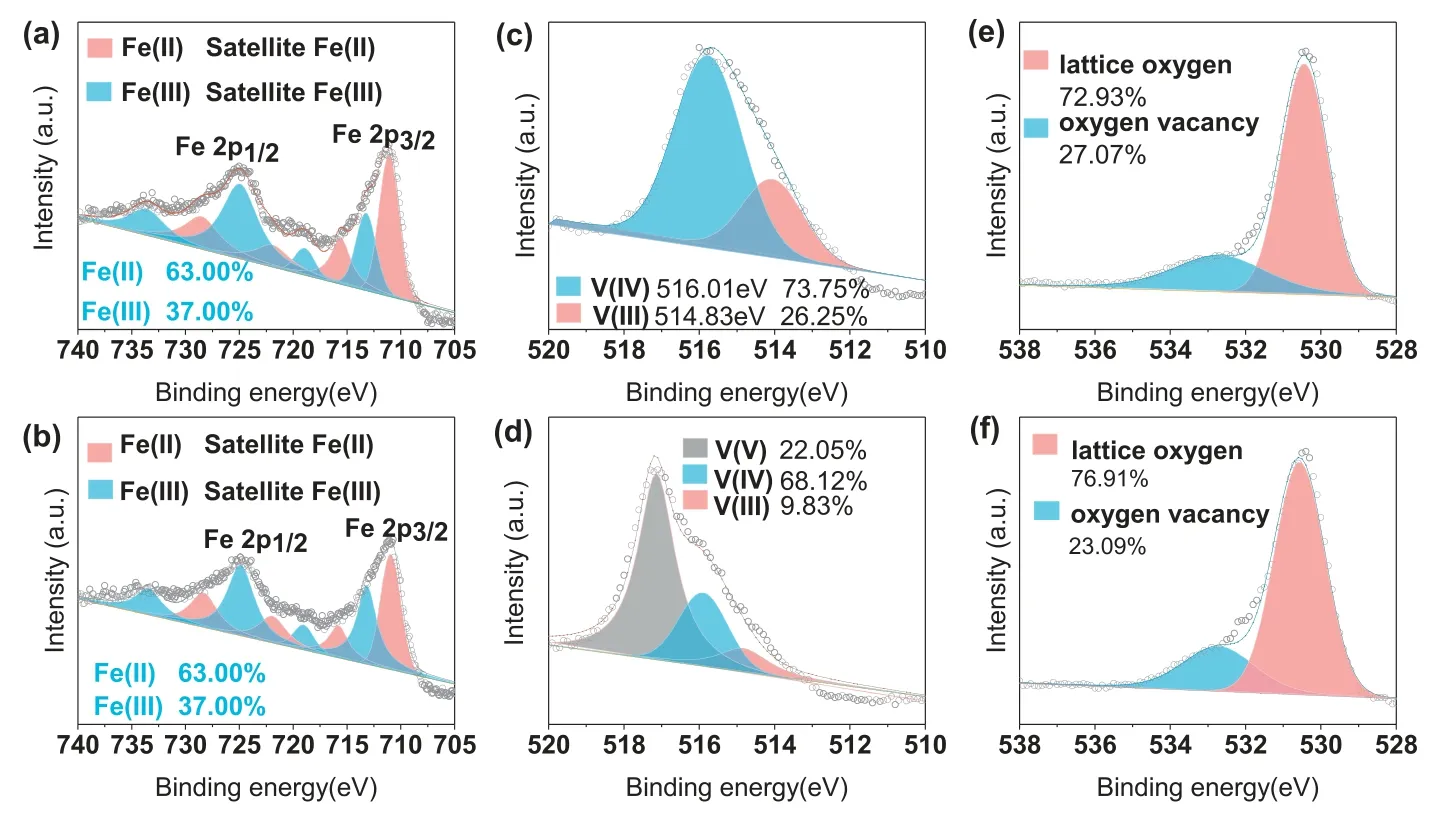
Fig.3.XPS spectra of fresh and used FeVO3-x: Fe 2p core level in the (a) fresh and (b) used FeVO3-x,V 2p core level in the (c) fresh and (d) used FeVO3-x,O 1s core level in the (e) fresh and (f) used FeVO3-x.
The electron paramagnetic resonance (EPR) test using 5,5-dimethyl-1-pyrrolineN-oxide (DMPO) as thein situradical spin-trapping reagent was conducted to qualitatively analyze the existence of the main ROS in the FeVO3-x/PMS system.In contrast to PMS alone and FeVO3-xalone systems (Fig.2b),two signals appearing in the FeVO3-x/PMS system are assigned to DMPO-HO adduct (four characteristic peaks with height ratio of 1:2:2:1,αN=αH=14.9 G) and DMPO-SO4adduct (αN=13.8 G,αH=10.1 G,αH=1.4 G,andαH=0.8 G) [18–20],implying both hydroxyl radicals (•OH) and sulfate radicals (SO4•-) were generated in the FeVO3-x/PMS system.However,since DMPO-SO4adduct is easily converted into DMPO-HO adduct [21],the signal intensity of DMPO-SO4adduct is weak in the FeVO3-x/PMS system.To further disclose the identity of•OH and SO4•-in the FeVO3-x/PMS system,tert-butyl alcohol (TBA) was introduced for•OH quenching(: 4.0–9.1 × 105L mol-1s-1,: 3.8–7.6 × 108L mol-1s-1) [22–24],and ethanol (EtOH) was introduced for both•OH and SO4•-quenching (: 1.6 × 107L mol-1s-1,: 1.9 × 109L mol-1s-1) [22].Fig.2c suggests that the quenching effects of EtOH increase as a function of concentration(50-200 mmol/L),while TBA has a feeble quenching effect on CBZ degradation regardless of the concentration (50-200 mmol/L).Besides,the semi-quantitative analysis of•OH in the FeVO3-x/PMS system were conducted with the terephthalic acid (TPA),since 2-hydroxyterephthalic acid (HTPA) as the characteristic product can be produced after the attack of•OH [25].Surprisingly,we found that the degradation trend of CBZ in the FeVO3-x/PMS system was similar to that in the classic co-catalytic Fenton system (hydroxylamine/Fenton,Fig.S4a in Supporting information).The ROS in the hydroxylamine/Fenton system are undoubtedly•OH.Therefore,the contribution of•OH and SO4•-formed in the FeVO3-x/PMS system can be better investigated by comparing with the hydroxylamine/Fenton system.Clearly,the generation concentration of HTPA in the FeVO3-x/PMS system was much lower (Fig.S4b in Supporting information) when the degradation trend of CBZ in the FeVO3-x/PMS system was similar to that in the classic co-catalytic Fenton system (hydroxylamine/Fenton).The collective results indicate that though both•OH and SO4•-are generated in the FeVO3-x/PMS system,the amounts of SO4•-are much more than that of•OH.
Since some studies pointed out that high-valent iron-oxo species (Fe(IV)) might be produced in the Fe mediated Fentonlike systems and the reaction rate between Fe(IV) and ethanol(kEtOH/Fe(IV): 2.51 × 103L mol-1s-1) is obviously faster than that between Fe(IV) and TBA (kEtOH/Fe(IV): 6 × 101L mol-1s-1)[19,26,27],the existence of Fe(IV) in FeVO3-x/PMS system was further qualitatively analyzed by the chemical probe method.Methyl phenyl sulfoxide (PMSO) was applied as a chemical probe because the reaction between PMSO with Fe(IV) proceeds through an oxygen atom transfer and methyl phenyl sulfone (PMSO2) is quantitatively generated [27].From the HPLC chromatograms,it is obvious that the concentration of PMSO was decreased while PMSO2was accumulated in the FeVO3-x/PMS system (Fig.2d).We further quantified the concentrations of PMSO2and PMSO to confirm the existence of Fe(IV).Results show that conversion efficiency of PMSO to PMSO2is 68.2% (Fig.2e),implying the role of Fe(IV) in the FeVO3-x/PMS system for CBZ degradation cannot be ignored.Moreover,some researchers argued that high-valent iron-oxo species might induce the production of O2•-[28,29].Therefore,EPR test was conducted with DMPO in the dimethyl sulfoxide (DMSO)solution to identify the existence of O2•-.The EPR spectrum with hyperfine splitting parameters ofαH=14.25 G,αN=12.45 G,and an intensity ratio of 1:1:1:1 is indexed to DMPO-O2adduct (Fig.2f)[30],indicating high-valent iron-oxo species reacting with PMS induces the formation of O2•-.Combined with the results of quenching experiment,although O2•-is produced in the FeVO3-x/PMS system,O2•-has a negligible effect on CBZ degradation.
To further elucidate the generation of ROS in the FeVO3-x/PMS system,the chemical compositions of FeVO3-xwere scrutinized by XPS analysis.The spectra of Fe 2p core level in the pristine and reacted FeVO3-xshow that the ratio of Fe(III) to Fe(II) is nearly invariable Figs.3a and b),indicating the generated Fe(III) induced by the reaction between PMS and Fe(II) can be quickly reduced during treatment process.In contrast with the chemical state of Fe,pronounced changes occurred in the V species after the final reaction (Figs.3c and d),where a new peak located at 517.20 eV assigned to V(V) [31,32] was found.The oxidation of V species in the FeVO3-x/PMS system follows two pathways: (i) low valent V species (V(III) and V(IV)) react with PMS (HSO5-) to generate V(V) (Eqs.1 and 2);(ii) low valent V species as electron donators transfer electrons to Fe(III) for fast Fe(II) regeneration and finally convert to V(V) (Eqs.3 and 4).The first pathway is confirmed by Fig.2a since 32% and 25% CBZ can be degraded in the V2O3/PMS and V2O4/PMS systems,respectively.The second pathway of low valent V species for Fe(III) reduction is further scrutinized in the following content.In addition,compared with fresh FeVO3-x,the decrease of oxygen vacancy in the used FeVO3-x(Figs.3e and f)also verifies that oxygen vacancy participated in the CBZ degradation.
To further investigate the role of low valent V species for Fe(III)reduction,the electronic structure and coordination environment of Fe and V species in the FeVO3-x,along with some other Fe and V-based oxides (FeO,Fe3O4,Fe2O3,V2O5,V2O4,and V2O3standards) were evaluated by X-ray absorption spectroscopy (XAS).E0was calculated as the maximum peak energy of the first derivative of the spectrum,which was used to identify the Fe oxidation state of different materials.According to the Fe K-edge X-ray absorption near edge structure (XANES) of FeVO3-xand other Fe-based oxides(Fig.4a),the value (7112.2 eV) ofE0in FeO spectrum is lowest,while in Fe2O3spectrum is highest (7114.1 eV),indicatingE0in Fe K-edge XANES spectrum increases as a function of chemical valence of Fe.Since the value (7112.7 eV) ofE0in the FeVO3-xis between that of FeO and Fe2O3,both Fe(II) and Fe(III) exist in the FeVO3-x.However,only Fe(III) exists in the FeVO3-xprecursor because itsE0is the same as that of Fe2O3.In addition,in contrast with Fe3O4,the pre-edge peak intensity of FeVO3-xis weaker than that of Fe3O4,implying the amount of Fe(II) in the FeVO3-xis more than that of in the Fe3O4,since the 3d orbital of Fe(II) possesses less electron number than that of Fe(III),and the pre-edge peak represents the 1s →3d electron transition [33].

Fig.4.(a) Fe K-edge XANES analysis,(b) V K-edge XANES analysis,FT curves of (c) Fe K-edge and (d) V K-edge EXAFS k3χ(k) functions obtained from the XANES spectra without fitting.(e,f) The fitting values of V K-edge EXAFS k3χ(k) functions obtained from the XANES spectra,and (g) WT of V-foil,FeVO3-x and FeVO3-x precursor.
As for the valence of V,the FeVO3-xprecursor,V2O5,V2O4,and V2O3standards have a pre-edge peak with prominent intensity,which drastically increases with V species oxidation states(Fig.4b).However,the pre-edge peak pattern of FeVO3-xis in remarkable contrast to those of other V-based materials.Since the symmetry of the V species in FeVO3-xdiffers from FeVO3-xprecursor,V2O3,V2O4,and V2O5,the oxidation states of V species in the FeVO3-xcannot be defined only by the pre-edge peak intensity.On the other hand,the V K-edge XANES spectrum of FeVO3-xis in line with the literature reports [4,34],indicating the oxidation state of V species in the FeVO3-xis trivalent and tetravalent.
The Fourier-transformed (FT) extended X-ray absorption fine structures of Fe K-edge (EXAFSk3χ(k)) are shown in Fig.4c.The peak at ∼1.5 ˚A (without phase correction) can be identified as Fe-O bond.Comparing FeVO3-xprecursorversusFeVO3-x,the peak at ∼1.5 ˚A shifts to much lower intensity in FeVO3-x(Fig.4c),suggesting the coordination number decreases in FeVO3-x,which further well-documents the existence of oxygen vacancies.Since the FT curves of V K-edge EXAFSk3χ(k) functions (Fig.4d) are more complicated than that of Fe K-edge EXAFSk3χ(k),the fitting curves of FeVO3-xcompared with FeVO3-xprecursor and V foil are shown in Fig.4e,Fig.4f and Table S1 (Supporting information).The well-resolved peaks at ∼1.4 ˚A and ∼3.0 ˚A (without phase correction) in FeVO3-xand FeVO3-xprecursor are indexed to V-O bond and V-O-Fe bond,respectively.The coordination numbers of V-O and V-O-Fe shells in the FeVO3-xare 3.0 and 0.6,which is in marked contrast to that of in the FeVO3-xprecursor(5.6 and 3.0,respectively,Table S1),also indicating the existence of existence of oxygen vacancies.Moreover,the wavelet transform plots (Fig.4g) of FeVO3-xand FeVO3-xprecursor also reflect the existence of V-O and V-O-Fe bonds.The existence of V-O-Fe bond facilitates the interpretation of the interelectronic interaction of Fe and V species in FeVO3-x.Specifically,the electron-riched t2gdorbitals of V species donating electrons to the electron-deficient t2gd-orbitals of Fe(III) species through the bridging O2-via πdonation [4].Moreover,CBZ degradation and Fe(II) generation after external addition of Fe(III) into V2O4/PMS and V2O3/PMS systems (Fig.S5 in Supporting information) also imply low-valent V species as electron-rich promoters mediate electron shuttles to expedite Fe(III) reduction and pollutant degradation.To further confirm the interaction of oxygen vacancy and PMS molecules,we further conductedin-situRaman spectroscopy analysis.Thein situRaman spectroscopy shows that the peak intensities of FeVO3-xat 146,286,and 703 cm-1increase after adding PMS (Fig.S6 in Supporting information),indicating peroxo species bound to the surface oxygen vacancies might be formed [17].
Previous studies suggested that co-existing anions in water might react with•OH and SO4•-(Eqs.5-12) [35–37].Therefore,the effects of different anions,such as Cl-,H2PO4-,NO3-and HCO3-,on CBZ degradation in the FeVO3-x/PMS system were investigated.From Fig.5a,it is clear that the common anions,such as Cl-,H2PO4-and NO3-in natural water almost show negligible effects on CBZ degradation,which suggests the great matrix resistance of this system.The obvious inhibition of CBZ degradation in the presence of HCO3-might originate from the considerable alkalinity induced by the introduction of HCO3-.However,in actual water samples such as tap water,Jinjiang water and Jiangan water,the degradation performance severely decreased (Fig.5b).This might be ascribed to the pH buffer effects of natural water samples and the presence of natural organic matters,which could severely compete with target contaminants for oxidative ROS.Finally,the system exhibited satisfactory cyclic performance during consecutive runs (Fig.5c).Despite a little decrease of CBZ removal from 100% to 80%,a facile N2or NH3regeneration could effectively restore the reactivity of used FeVO3-xand ensure the long-term stability.
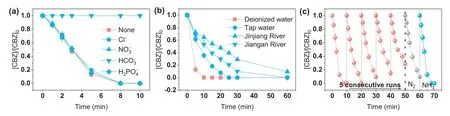
Fig.5.(a) The effect of Cl-,NO3-,HCO3-,and H2PO4- co-existing ions on CBZ degradation.(b) CBZ degradation in different water samples.(c) CBZ degradation in FeVO3-x/PMS system during 5 consecutive runs and after FeVO3-x regeneration.Experiment condition: [FeVO3-x]0=100 mg/L,[PMS]0=0.2 mmol/L,[CBZ]0=2.5 mg/L,pHini=6.3,[Cl-]=5 mg/L,[NO3-]=5 mg/L,[HCO3-]=20 mg/L,[H2PO4-]=5 mg/L.
Based on the detected seven intermediates,we proposed the possible degradation pathway of CBZ in the FeVO3-x/PMS system(Fig.6a,Table S2 and Figs.S7-S14 in Supporting information).First,the olefinic unsaturated bond of central heterocyclic ring is readily attacked [4,38],and thusP1(m/z=251.0821) andP2(m/z=269.0922) are formed owing to the attack of SO4•-,•OH and Fe(IV).P2is then oxidized toP5(m/z=267.0772)viaintramolecular cyclisation and carboxylation reactions.In addition,the dialdehyde moieties ofP2are unstable,which leads to the rotation of benzene ring ofP2to generateP6(m/z=267.0774).The aldehyde moiety on theP6can be further oxidized by the ROS to form a carboxylic acid product (P7,m/z=283.0723).Subsequently,P7could be oxidized to form P4 (m/z=196.0766)viadeamination,acrylamido abstraction,and decarboxylation reactions.Moreover,P1can also be oxidized to generateP3(m/z=180.0813)viaintramolecular cyclisation,amine/acrylamido cleavage and de-formyl,which further be oxidized to produceP4.Finally,these detected intermediates are mineralized to CO2and H2O.Specifically,the TOC removal drastically increased with treatment time (Fig.S15 in Supporting information): the TOC removal reached 17% after 10 min treatment,while it increased to 55% after 120 min treatment.
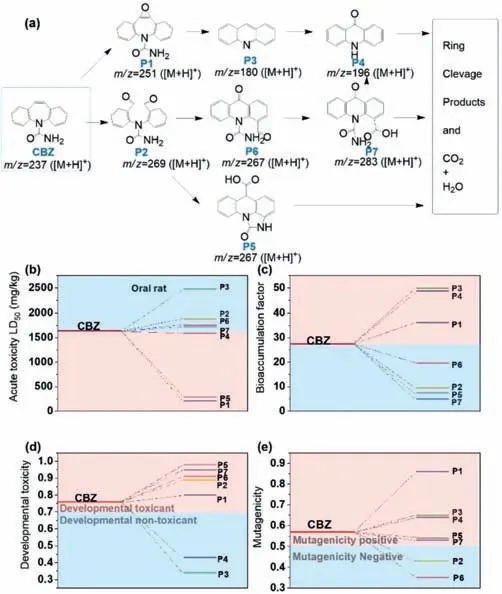
Fig.6.(a) CBZ degradation pathways in the FeVO3-x/PMS system.(b) Acute toxicity,(c) bioaccumulation factor,(d) developmental toxicity,and (e) mutagenicity of CBZ and its intermediates.Experimental conditions for (a): [FeVO3-x]0=100 mg/L,[PMS]0=0.2 mmol/L,[CBZ]0=2.5 mg/L,and initial pHini=6.3.
Furthermore,we also evaluated the developmental toxicity,acute toxicity,bioaccumulation factor and mutagenicity of original CBZ and its intermediates through the Toxicity Estimation Software Tool.Fig.6b shows the oral rat LD50 ofP1andP5are 213.16 and 287.73 mg/kg,respectively,which are much lower than that of CBZ(1636.63 mg/kg),implying the acute toxicities ofP1andP5are quite more toxic than original CBZ.However,the acute toxicities of most types of intermediates are much lower than CBZ.Since the potential hazards of these intermediates are negative correlation with bioaccumulation factor,and Fig.6c shows the bioaccumulation factors ofP2,P5,P6,P7(9.56,7.41,19.70,and 4.98,respectively) are much lower than that of CBZ (27.38),four intermediates have quite lower risks than CBZ.However,the results of developmental toxicity (Fig.6d) and mutagenicity (Fig.6e) suggest that five developmental toxicants (P1,P2,P5,P6andP7) and five mutagenicity negative intermediates (P1,P3,P4,P5andP7) are generated after treatment process.
In this work,we synthesized a novel catalysts FeVO3-xviaa facile ammonia reduction method.The high-temperature reduction process endowed the catalysts with abundant low-valent V species and electron-rich oxygen vacancy,which are both conducive to the circulation of Fe(III) to Fe(II).Through quenching experiments and EPR analysis,we found that•OH,SO4•-,O2•-and Fe(IV) were all generated in this system,but only SO4•-and Fe(IV) primarily contributed to the degradation of CBZ.X-ray photoelectron spectroscopy and X-ray adsorption spectroscopy indicated that both the low-valent V provides and oxygen vacancy could accelerate the internal electron transfer to Fe(III)viathe unsaturated V-O-Fe bond.Despite a little decrease of performance after consecutive runs,the activity could be effectively regeneratedviare-calcination treatment.Finally,the degradation product and the corresponding toxicity analysis revealed that this system is basically a toxicity attenuation process,demonstrating its potential in practical application.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The first author is funded by the Shanghai Tongji Gao Tingyao Environmental Science &Technology Development Foundation.Additionally,the authors acknowledge the staff at beamline 1WB at the Beijing Synchronic Radiation Facility (BSRF) for their assistance during the XAS measurements.And the authors would like to acknowledge the financial support from National Natural Science Foundation of China (Nos.52070133,2022NSFSC0972),Sichuan Science and Technology Program: Key Research and Development Program (Nos.2019YFG0314,2017SZ0180 and 2019YFG0324).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108580.
杂志排行

Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers