Low-temperature selective synthesis of metastable α-MoC with electrochemical properties: Electrochemical co-reduction of CO2 and MoO3 in molten salts
2024-04-06LongtoZhuYinnZhoWenhoYngHsienYiHsuPingPengFngFngLi
Longto Zhu ,Yinn Zho ,Wenho Yng ,Hsien-Yi Hsu ,Ping Peng,∗ ,Fng-Fng Li,∗
a School of Materials Science and Engineering,Huazhong University of Science and Technology,Wuhan 430074,China
b School of Energy and Environment &Department of Materials Science and Engineering,City University of Hong Kong,Hong Kong 999077,China
Keywords: α-MoC CO2 electroreduction Molten salt Sulfur doping Hydrogen evolution reaction
ABSTRACT Metastable molybdenum carbide (α-MoC),as a catalyst and an excellent support for metal catalysts,has been widely used in thermo/electro-catalytic reactions.However,the selective synthesis of α-MoC remains a great challenge.Herein,a simple one-pot synthetic strategy for the selective preparation of metastable α-MoC is proposed by electrochemical co-reduction of CO2 and MoO3 in a low-temperature eutectic molten carbonate.The synthesized α-MoC shows a reed flower-like morphology.By controlling the electrolysis time and monitoring the phase and morphology of the obtained products,the growth process of α-MoC is revealed,where the carbon matrix is deposited first followed by the growth of α-MoC from the carbon matrix.Moreover,by analyzing the composition of the electrolytic products,the formation mechanism for α-MoC is proposed.In addition,through this one-pot synthetic strategy,S-doped α-MoC is successfully synthesized.Density functional theory (DFT) calculations reveal that S doping enhanced the HER performance of α-MoC by facilitating water absorption and dissociation and weakening the bond energy of Mo-H to accelerate H desorption.The present work not only highlights the valuable utilization of CO2 but also offers a new perspective on the design and controllable synthesis of metal carbides and their derivatives.
Transition metal carbides (TMCs),owing to their high electrical conductivity,excellent durability as well as wide pH applicability,have attracted great attention in the materials science community [1–4].Among them,molybdenum carbides possess similar d-band electronic structures to platinum [5,6],which makes them widely used in catalytic reactions such as ammonia preparation and decomposition [7–9],steam reforming of methanol [10,11],water-gas shift (WGS) reaction [12–14],and electrochemical hydrogen evolution reaction (HER) [15–19].Besides,molybdenum carbides can also be used for energy storage [20,21].The hexagonalclose-packedβ-Mo2C and the metastable face-centered-cubicα-MoC are the most studied molybdenum carbide phases.Compared withβ-Mo2C,α-MoC has a d-band center closer to the Fermi level,which is favorable for the adsorption and dissociation of water molecules,thus,α-MoC-based catalysts exhibited higher activity thanβ-Mo2C-based catalysts in methanol steam reforming,WGS reaction,and alkaline HER [11,12,22].In addition,α-MoC ex- hibits stronger interactions with metals thanβ-Mo2C,so it has been used as a support for metal species with catalytic activity.A series of high-performance M/α-MoC (M=Pt,Ru,Ir,Au,and Ni)heterogeneous catalysts have been established.Due to the strong metal-support interaction,α-MoC not only facilitated the stabilization of metal single atoms and/or metal clusters but also promoted the catalytic activity of the metal centersviaoptimizing adsorption/desorption of the reactants and intermediates on the catalysts surface [10–13,17,23,24].
Nevertheless,compared with the thermally stableβ-Mo2C phase,the selective synthesis of the pure metastableα-MoC phase remains challenging because of the complex procedures,noble metal requirement,and high energy input [22,25–27].The current synthesis method forα-MoC mainly relies on the high-temperature carbonization/carburization of Mo-containing precursors,which often cause the aggregation and coked surfaces ofα-MoC to prevent the exposure of active sites [17,28–31].Moreover,the usage of combustible/explosive gas reductant,i.e.CH4/H2forα-MoC is highly dangerous [26,27,32–34].In addition,the carbon sources for the preparation ofα-MoC are usually environment-unfriendly organic compounds [23,24,35].Thus,given the highly valuable applications ofα-MoC and the present harsh synthetic conditions,a simple low-temperature process to selectively synthesize theα-MoC phase is urgently needed.
Carbon dioxide (CO2) is a predominant greenhouse gas but is also an abundant and cost-effective carbon feedstock for producing high-value carbon materials.In molten salt media,CO2can be efficiently captured [36,37] and electrochemically converted into highly valuable nanocarbons [38–44] and metal-carbon composites [45–48].Molten salts bear low toxicity,low cost,and high ionic conductivity.Moreover,molten media has wide electrochemical and temperature windows (200-1000◦C),which can be tuned by changing the composition of the molten salts as well as the proportion of the salts.Therefore,using CO2as the carbon source for the selective synthesis ofα-MoC in the low-temperature molten salt is a low-cost sustainable approach,which is expected to address the aforementioned issues associated with the low-selectivity and harsh synthesis conditions.In addition,the growth process and formation mechanism ofα-MoC in molten media are rarely studied.Understanding the reaction process and kinetics is important for the design and synthesis ofα-MoC-based/supported materials.
In this work,we propose a simple method for selective synthesis of metastableα-MoC phase by electrochemical co-reduction of CO2and MoO3in molten carbonates at low temperatures (500-600◦C).The as-synthesizedα-MoC shows a reed flower-like morphology composed of ultrafine MoC particles.The growth process forα-MoC was revealed by controlling the electrolysis time and monitoring the as-obtained samples byex-situXRD and TEM.The formation mechanism was unraveled by analyzing the composition of the electrolytic products.In addition,by introducing Li2SO4into the molten carbonate electrolyte,S-dopedα-MoC was synthesized in one step.S-dopedα-MoC exhibits enhanced alkaline HER activity relative toα-MoC,which was rationalized by density functional calculations (DFT).The present work not only provides a new perspective for understanding the formation ofα-MoC but also offers an alternative approach to the synthesis of metal carbides and their derivatives.
As illustrated in Scheme 1,the eutectic carbonate (Li2CO3,Na2CO3,and K2CO3) and molybdenum source (MoO3) are melted at different temperatures (500◦C,550◦C,and 600◦C) to serve as the electrolyte.A graphite rod and a galvanized steel with a springlike shape are used as the anode and the cathode (see photographs of the cathode before and after electrolysis in Fig.S1 (Supporting information),respectively.After a constant current of 0.5 A is applied for 1 h,α-MoC is deposited on the surface of the cathode.The corresponding electrolysis curves are shown in Fig.S2 (Supporting information).The electrolytic products are washed with acid and deionized water to obtain theα-MoC products.α-MoC obtained at different temperatures are namedα-MoC-500,α-MoC-550,andα-MoC-600,respectively.
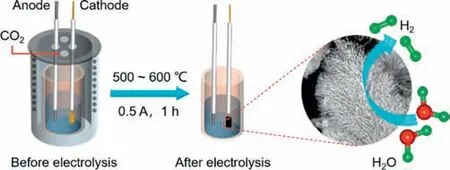
Scheme 1.Schematic illustration for the synthesis of α-MoC or S-doped α-MoC in molten salts.
The structures of the synthesized samples were first characterized by X-ray diffraction (XRD).As shown in Fig.1a,four peaks located at 36.4°,42.3°,61.4°,and 73.5°,respectively correspond to the (111),(200),(220),and (311) planes ofα-MoC (PDF# 89-2868) [28],indicating the successful synthesis ofα-MoC.To analyze the surface chemical constituent of the samples,X-ray photoelectron spectroscopy (XPS) was performed.The survey spectra(Fig.S3 in Supporting information) show thatα-MoC obtained at different temperatures consists of C,Mo,and O elements.The highresolution XPS spectra of Mo 3d can be deconvoluted into eight peaks (Fig.1b and Table S1 in Supporting information),which represent the four valence states (+2,+3,+4,and +6) of the Mo element.The peaks of Mo2+and Mo3+are associated with the Mo-C bond,and the peaks of Mo4+and Mo6+correspond to MoO2and MoO3due to the surface oxidation of the samples in the air[47,48],which accounts for the presence of O element in the full XPS survey (Fig.S3).Besides,the C 1s spectra ofα-MoC were also analyzed,and there are four peaks in the C 1s spectrum,representing the C-Mo,C-C,C-O,and C=O bonds (Fig.1c and Table S2 in Supporting information) [49,50].
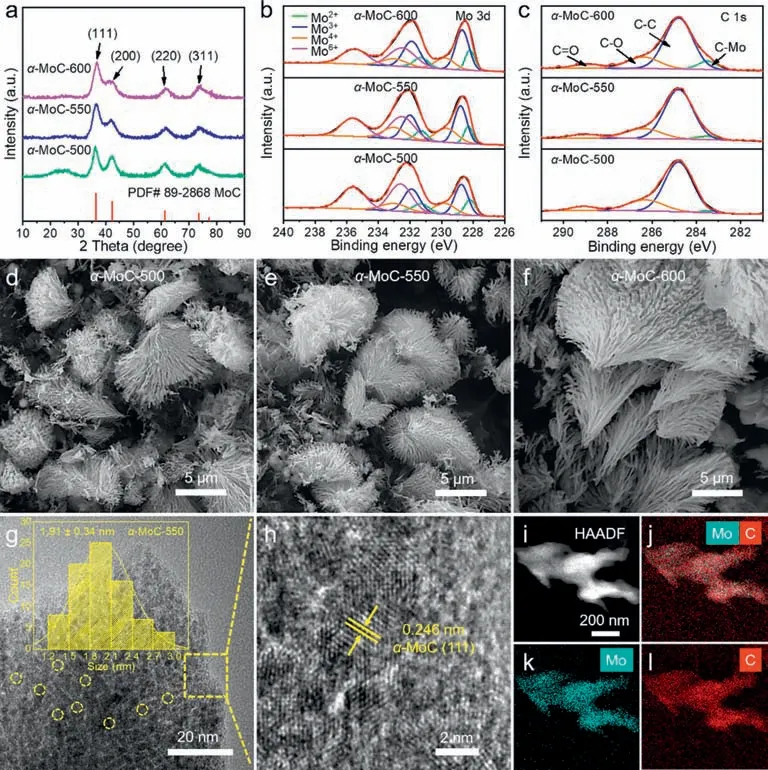
Fig.1.(a) XRD patterns,(b,c) the high-resolution XPS spectra of Mo 3d and C 1s,(d–f) SEM images of the synthesized α-MoC at different temperatures.(g,h) HR-TEM images,(i–l) HAADF-STEM and the corresponding EDS elemental mapping images of α-MoC-550.
The morphology of the samples was examined by scanning electron microscopy (SEM).α-MoC prepared at different temperatures showed a reed flower-like morphology (Figs.1d–f).TEM analysis of the representativeα-MoC-550 indicated that each branch of reed flowers consists of many small particles with a size distribution of 1.91 ± 0.34 nm (Fig.1g).The lattice fringes of the particles have an interplanar spacing of 0.246 nm,corresponding to the(111) plane ofα-MoC (Fig.1h),which indicates thatα-MoC particles are well crystallized at 550 °C.Moreover,a uniform distribution ofα-MoC nanoparticles was observed from the HAADF-STEM and the corresponding EDS elemental mapping images ofα-MoC-550 (Figs.1i–l).As the electrolysis temperature increases to 600 °C,α-MoC-600 presents larger grains with a size of 2.23 ± 0.27 nm(Figs.S4a and b in Supporting information).Whereas,α-MoC-500 obtained at a low temperature of 500 °C mainly shows an amorphous structure with only crystallization in local areas (Figs.S4c and d in Supporting information).
In order to study the growth process ofα-MoC,we conducted control experiments by controlling the electrolysis time and monitored the reaction byex-situXRD and TEM.The electrolysis was carried out at 550◦C Li2CO3/Na2CO3/K2CO3electrolyte for 5 min,10 min,15 min,and 20 min,and the corresponding products are namedα-MoC-550-5,α-MoC-550-10,α-MoC-550-15,andα-MoC-550-20,respectively.XRD patterns for the products were plotted in Fig.2a.When the electrolysis time is controlled at 5 min,the XRD pattern ofα-MoC-550-5 is mainly the diffraction peaks of carbon.With the extension of electrolytic time from 5 min to 10 min,15 min,and 20 min,the carbon peaks gradually decreased and the characteristic peaks ofα-MoC are correspondingly enhanced,indicating the gradual conversion of carbon into MoC.
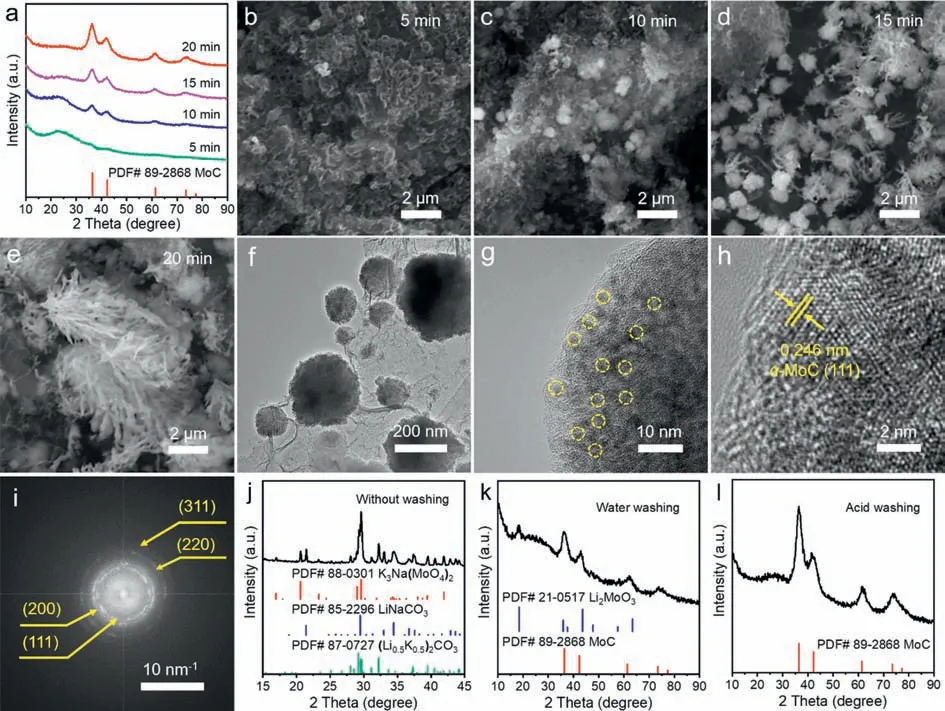
Fig.2.(a) XRD patterns of the samples obtained at electrolysis times of 5 min,10 min,15 min,and 20 min,respectively.(b–e) SEM images of α-MoC-550-5,α-MoC-550-10,α-MoC-550-15,and α-MoC-550-20.(f) TEM image of α-MoC-550-10.(g,h) HR-TEM images and (i) the corresponding selected-area electron diffraction pattern (SAED) of α-MoC-550-10.XRD patterns of the electrolytic products (j) without washing,(k) with water washing,and (l) with acid washing.
SEM images of the products obtained at different times are shown in Figs.2b–e.The carbon obtained by 5 min electrolysis is amorphous (Fig.2b),and then some spherical particles start to grow out from the carbon matrix (Fig.2c).Interestingly,when the electrolysis time was extended to 15 min,some branches start to grow out from the surface of spherical particles (Fig.2d).At 20 min,α-MoC with reed-flower like morphology was formed(Fig.2e).To identify the structure of the spherical particles in Fig.2c,TEM analysis was performed,which showed that the spherical particles attached to the carbon substrate (Fig.2f).HR-TEM and the corresponding selected-area electron diffraction pattern (SAED)revealed that the spherical particle is composed of smallerα-MoC crystalline grains with a lattice space of 0.246 nm,corresponding to the (111) plane ofα-MoC (Figs.2g–i).The diffraction rings from inside to outside represent the (111),(200),(220),and (311) planes ofα-MoC,respectively (Fig.2i).Based on the above analysis,we proposed the formation process ofα-MoC as follows: The carbon substrate was first deposited on the cathode surface,and then the sphericalα-MoC grains are grown out from the carbon substrate and served as the nucleation sites forα-MoC growth.
To unravel the formation mechanism ofα-MoC,we analyzed the composition of the electrolytic products which were treated with different washing conditions.The cathodic product without washing presents a color grey (Fig.S5 in Supporting information)and an irregular morphology (Fig.S6a in Supporting information)since the electrolyte is wrapped around the product.The XRD pattern of the unwashed product showed the characteristic diffraction of LiNaCO3and (Li0.5K0.5)2CO3(Fig.2j) due to the formation of new carbonates by melting,cooling,and recrystallization of Li2CO3,Na2CO3,and K2CO3.Notably,the diffraction peaks of K3Na(MoO4)2were also observed in the XRD spectrum and K3Na(MoO4)2served as the Mo source for the formation ofα-MoC.Unfortunately,due to the strong diffraction peaks of the electrolyte,the signals ofα-MoC were masked.When the electrolytic product was washed with deionized water,the sample appeared black (Fig.S5),indicating that most of the carbonate electrolytes were eluted,but its morphology remained heterogeneous (Fig.S6b in Supporting information).Consistent results were obtained from its XRD analysis.The diffraction peaks of LiNaCO3,(Li0.5K0.5)2CO3,and K3Na(MoO4)2disappeared and the diffraction peaks ofα-MoC and Li2MoO3were observed in this water-washed product (Fig.2k).The appearance of Li2MoO3is owing to the cathodic reduction of MoO42-to MoO32-,which combines with Li+in the electrolyte to form the waterinsoluble Li2MoO3.Further,when the electrolytic product is treated with acid,the product showed black color (Fig.S5) and a homogeneous reed-flower morphology (Fig.S6c in Supporting information),indicating that the carbonate electrolyte was completely removed.Consistent results were obtained from the XRD spectrum,in which only the diffraction peaks ofα-MoC were presented(Fig.2l).
Notably,K3Na(MoO4)2was detected in the electrolytic product (Fig.2j).To understand the formation of MoO42-,tail gases evolved from MoO3-mixed Li/Na/K carbonates (without electrolysis) at room temperature and 550◦C (without electrolysis) were analyzed under Ar gas flow.As displayed in Figs.S7a and b(Supporting information),the volume of O2collected at room temperature (0.44%) and 550◦C (0.48%) was almost the same.As the temperature increased from room temperature to 550◦C,the CO2content sharply increased from 0.27% to 13.02%,which indicated that the MoO42-came from the spontaneous reaction between MoO3and carbonate (MoO3+CO32-→MoO42-+CO2) at 550◦C.Furthermore,the composition of tail gases before and after the electrolysis under CO2atmosphere at 550◦C was analyzed (Figs.S7c and d in Supporting information).The O2content increased from 0.72% to 2.42%,and the CO2content decreased from 90.83%to 87.14%,suggesting the capture and conversion of CO2as well as the formation of O2during the electrolysis process.
Based on the above results,the formation mechanism ofα-MoC by molten salt electrolysis can be proposed as follows: MoO3reacted with CO32-to form MoO42-and CO2(Eq.1).CO2was captured by O2-generated from the electroreduction of MoO42-(Eq.2) and/or CO32-splitting (Eq.3) to regenerate the CO32-(Eq.4).The electroreduction of MoO42-also produced the MoO32-(Eq.2),which was co-reduced with C generated from CO32-reduction (Eq.3) to formα-MoC (Eq.5).The generated O2-ions (Eqs.2 and 3) played two roles in this conversion process.Part of O2-was oxidized to O2by losing electrons at the anode (Eq.6),and the rest of O2-captured CO2to regenerate CO32-(Eq.4) [36,37].Thus,the net reaction is the co-reduction of MoO3and CO2to form theα-MoC and release the O2(Eq.7).The detailed reaction process can be found in Fig.S8 (Supporting information).The electrolytic effi-ciency forα-MoC synthesized at 500,550,and 600◦C is 63.18%,66.40%,and 72.56%,respectively (Table S3 in Supporting information).
In addition,we also performed the electrolysis in Li2CO3/Na2CO3/K2CO3electrolyte without adding MoO3,where a Mo anode was used instead of a graphite rod anode (Fig.S9 in Supporting information).As expected,the XRD pattern of the electrolytic product is consistent with that ofα-MoC (Fig.S10 in Supporting information).The SEM images of the obtained product also show a reed flower-like morphology (Fig.S11 in Supporting information),indicating the formation ofα-MoC.These results suggested that during the electrolysis,the Mo anode wasin-situoxidized to MoO3and served as the Mo source.Thus,α-MoC can be synthesized from CO2andin-situgenerated MoO3in one step.
Molybdenum carbides have been applied as electrocatalysts in hydrogen evolution reactions (HER),and heteroatom doping is considered to be effective to promote catalytic performance [28,51,52].Therefore,based on the preparation method ofα-MoC,we also synthesized heteroatom-dopedα-MoC.By adding Li2SO4into the 550◦C molten Li2CO3/Na2CO3/K2CO3,S-dopedα-MoC-550 was successfully obtained by one-step electrolysis.The XRD pattern(Fig.3a),SEM (Fig.3b),and TEM images (Fig.3c) showed that the structure and morphology of S-dopedα-MoC-550 are similar to that ofα-MoC-550.Namely,S-dopedα-MoC-550 shared the same reed flower-like morphology asα-MoC-550.S-dopedα-MoC-550 is also composed of small crystallineα-MoC particles with a size distribution of 1.87 ± 0.27 nm (Figs.3d and e).The successful doping and uniform distribution of S element in S-dopedα-MoC-550 was confirmed by its HAADF-STEM and the corresponding EDS elemental mapping (Figs.3f–i).Element contents of S-dopedα-MoC-550 according to EDS spectra were listed in Table S4 (Supporting information),where the content of S isca.3.49 wt%.The Mo 3d spectrum (Fig.3j and Table S5 in Supporting information) of Sdopedα-MoC-550 was also similar to that ofα-MoC-550 (Fig.1b).The C 1s spectrum (Fig.3k and Table S6 in Supporting information) of S-dopedα-MoC-550 can be split into five peaks,and the peak at 285.5 eV represents the C-S bond [53].The S 2p spectrum(Fig.3l and Table S7 in Supporting information) could be deconvoluted into five peaks,and the peak at 161.9 eV is associated with the S-Mo bond [54].The peaks located at 163.2 eV and 164.5 eV were assigned to the C-S-C bond,and another two peaks at 168.2 eV and 169.2 eV were ascribed to the presence of sulfone [55,56].The presence of the C-S bond (Fig.3k) and the S-Mo bond (Fig.3l)further elucidated the successful doping of S intoα-MoC.
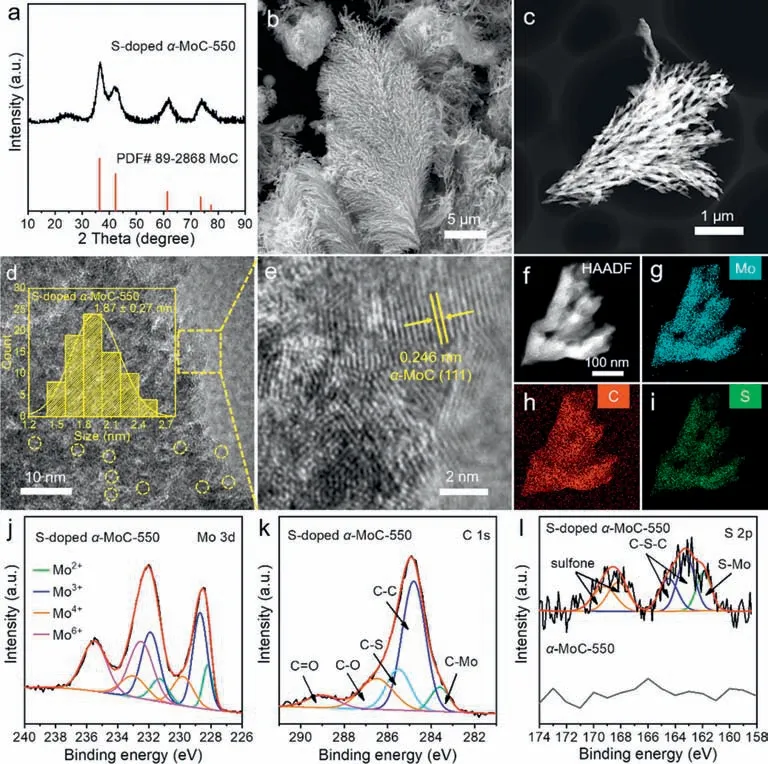
Fig.3.(a) XRD pattern,(b) SEM,(c) HAADF-STEM,and (d,e) HR-TEM images of the synthesized S-doped α-MoC-550.(f–i) HAADF-STEM and the corresponding EDS elemental mapping images of S-doped α-MoC-550.The high-resolution XPS spectra of (j) Mo 3d,(k) C 1s,and (l) S 2p for S-doped α-MoC-550.
To assess the electrocatalytic performance ofα-MoC and Sdopedα-MoC toward HER,linear sweep voltammetry (LSV) was performed in 1 mol/L KOH (Fig.4a).S-dopedα-MoC performed better thanα-MoC-550 for HER by showing a lower overpotential relative toα-MoC-550 at the same current density,which demonstrated the positive effect of S-doping on HER.Moreover,S-dopedα-MoC-550 andα-MoC-550 show lower overpotentials than Pt/C at current densities larger than 133 and 149 mA/cm2,respectively.The kinetics of HER was evaluated by calculating the Tafel slope (Fig.4b),where the Tafel slope of S-dopedα-MoC-550 is 80.2 mV/dec,smaller than that ofα-MoC-550 (83.6 mV/dec),indicative of the fast HER kinetics of S-dopedα-MoC-550 and a Volmer-Heyrovsky process [57].The transport kinetics of electrodes was studied by electrochemical impedance spectroscopy (EIS).The Nyquist plot of S-dopedα-MoC-550 shows a smaller semicircle thanα-MoC-550 (Fig.4c),representing a fast electron transfer process in the 1 mol/L KOH electrolyte.Additionally,the time-dependent current density curve of S-dopedα-MoC-550 showed an ignored decline after the 10 h test compared withα-MoC-550 and Pt/C (Fig.4d),indicating that S-dopedα-MoC-550 has excellent stability.The enhanced activity of S-dopedα-MoC-550 was rationalized by density functional theory (DFT) calculations.The models of MoC (111) and S-MoC (111) (Fig.S12 in Supporting information) were constructed based on the TEM and XPS results (Figs.1h,3e and l).Compared with MoC (111),S-MoC (111)has higher absorption energy and lower dissociation energy for water molecules (Fig.4e) as well as an H absorption energy closer to zero (Fig.4f),which is beneficial to water absorption,dissociation,and H desorption [58].In addition,compared with MoC (111),the charge density around the Mo atom of S-MoC (111) is significantly reduced (Fig.S13 in Supporting information),which is attributed to the higher electronegativity of the S atom relative to the Mo atom,leading to the charge transfer from Mo to S,thus weakening the bond energy of Mo-H [29] and facilitating the recombination of reactive hydrogen intermediates to produce H2.
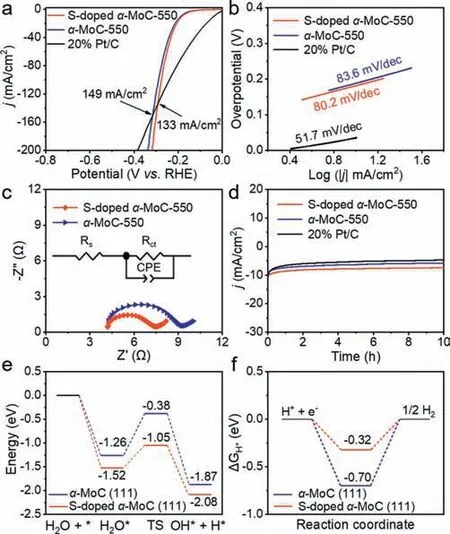
Fig.4.(a) Polarization curves at a scan rate of 5 mV/s with a 90% iR-compensation in 1 mol/L KOH.(b) Tafel slopes.(c) Nyquist plots at the overpotential of 150 mV.The inset is an electrical equivalent circuit model.(d) The time-dependent current density curve.(e) Adsorption and dissociation energy of the H2O molecule on the surface of α-MoC (111) and S-doped α-MoC (111).(f) Gibbs free energy diagrams of H adsorption on α-MoC (111) and S-doped α-MoC (111).
In summary,a low-temperature synthesis method forα-MoC was presented,in which CO2and MoO3were electrochemically coreduced in 500-600◦C molten carbonates.The growth process ofα-MoC suggested that the carbon substrate formed first followed by the growth ofα-MoC.Thein-situformation of MoO42-and the coupling reduction with CO2account for the formation ofα-MoC.Understanding the growth ofα-MoC will guide the design and synthesis ofα-MoC-based materials.In addition,based on the preparation method forα-MoC,S-dopedα-MoC was synthesized in one step by simply introducing Li2SO4into the electrolyte.The S doping enhanced the HER performance ofα-MoC by facilitating water absorption and dissociation and weakening the bond energy of Mo-H.This work not only demonstrated a facile way to prepare undoped/dopedα-MoC but also provided in-depth insight into the formation mechanism ofα-MoC.We propose that the strategy presented here could serve as a sustainable platform for the synthesis of other metal carbide materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
We appreciate the financial support from National Natural Science Foundation of China (Nos.22071070,21971077).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108583.
杂志排行

Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers