基于大数据技术的Cochran-Armitage算法的分布式研究与实现
2024-04-06吴双列王军凯杜江高洪睿
吴双列 王军凯 杜江 高洪睿

关键词:趋势检验算法;分布式;并行计算;大数据技术;计算集群
中图分类号:TP311.11 文献标识码:A
文章编号:1009-3044(2024)03-0078-07
0 引言
在基因组分析中,Cochran-Armitage趋势检验是非常重要的算法,其主要被用来完成基因型频率差异的统计检验[1-2]。Cochran-Armitage趋势检验在实际基因检测时,却存在两个问题:第一,每个生物体有大量基因信息,使用传统的Cochran-Armitage趋势检验算法进行计算,花费的时间成本会比较高。例如,一个人类样本可能包含超过4 000 000个染色体。如果有两个组,第一组有3 000个样本,第二组有5 000个样本。若要去比较两个组的基因型频率,必须分析32 000 000 000条记录。第二,Cochran-Armitage趋势检验算法每次只能对两个组进行计算,若同时有多个组要处理,则除正在运行的任务外,其余的处理任务可能因需要等待计算资源而无法及时处理。
针对以上两个问题,本文提出了基于大数据技术的分布式并行化Cochran-Armitage算法[3-8]。首先,本文使用分布式文件存储系统HDFS存储需要计算的数据并设计文件的读取逻辑。其次,设计算法的Map阶段以及Reduce阶段的处理逻辑;最后,实现了Spark 计算集群执行分布式并行化的计算任务。这种分布式的Cochran-Armitage趋势检验算法,不仅极大地缩减了运算时间,同时也降低了计算机资源的占用。
1 研究现状
Cochran-Armitage趋势检验算法常被应用在小样本和小批量的实验中。针对趋势检验的改进有两种方法:第一种是用其他的模型替换Cocharan-Armitage。例如2023年,Manning和Ku等人在遗传病例对照关联研究中使用了Jonchheere-Terpstra趋势检验[9],这是Cochran-Armitage趋势检验的非参数替代方法。第二种是将趋势检验与其他模型结合。例如2023年,Mesa和Analuisa等人在研究肉瘤患者合并症与住院死亡率之间的关联时将Cochran-Armitage趋势检验与多元二项式逻辑回归结合[10]。
对于大样本和大批量的应用场景,它们的效果并不太理想。传统的趋势检验算法都是串行实现,会消耗大量的时间并降低了工作效率。所以,文本对分布式并行化的Cochran-Armitage 算法展开了深入的研究。
2 Cochran-Armitage 分布式實现
2.1 Cochran-Armitage 趋势检验原理
20世纪下半叶,Cochran和Armitage提出并完善了Cochran-Armitage 算法[11-12],Cochran-Armitage 算法一经推出大受好评,被认为是基因科学中非常重要的一个计算指标。在基因组分析中,Cochran-Armitage 算法可以完成基因型频率(genotype frequency) 差异的统计检验,主要用来计算P值,P值越小则差异越大。Cochran-Armitage趋势检验根据基因数据为每个等位基因建立2行3列的列联表。列联表的行表示分组,列联表的列表示该等位基因在每条数据中出现相应计数的条数。
在表1和表2中,行标签(组A与组B) 表示基因组分析的两个不同组别,两个组的样本都是不相同的。列标签(0的计数、1的计数和2的计数)表示在某个组中一条基因数据里某个等位基因存在0、1或2个的样本数量。例如,组A有2个样本,其中一个样本的等位基因1为A,等位基因2为C;另一个样本的等位基因1 为A,等位基因2为A。则对于等位基因A的列联表中,组A中0的计数表示为0;1的计数表示为1;2的计数表示为1。表2中位于行标签和列标签的总和分别表示对某一行求和与对某一列求和。
得到列联表后,分别计算行和列的边缘总和,以及整个表的总和。
2.2 经典串行Cochran-Armitage 算法的分析
串行算法是单台机器单线程的处理任务。流程如图1。
如图1 所示。首先,初始化分布式文件系统HDFS既获取与分布式文件系统HDFS的连接,为读取数据做准备。接着,获取组A 与组B 数据文件(aGroup.txt 和bGroup.txt) 的输入流并更新列联表。最后,分别计算各个等位基因的P 值并输出结果。
1) 存在的问题
①问题1:图1中每个矩形都代表一个逻辑任务。对于串行计算,每一个任务都要等待上一个任务彻底的处理完成才可以开始。但是这些任务之间并不是完全依赖关系,对于获取aGroup.txt输入流读入数据切割并更新列联表的阶段和获取bGroup.txt输入流读取数据切割并更新列联表的阶段,这是两个完全不依赖彼此执行的阶段。可将两个阶段并行执行来解决,但串行任务处理的特性限制了这个方法。
②问题2:在读入数据切割并更新列联表阶段,对于处理大数据量问题,如百万数据、千万条甚至亿条数据,都是单机处理。如果一条数据处理用时0.05ms,那么1亿条数据就需要消耗5 000 000ms,这是非常大的资源消耗。如果将这些数据交由N台机器同时处理则可解决这个问题。每台机器所处理的数据量以及处理数据所消耗的时间均减少至原来的1/N倍。
上述2个问题在单机串行模式下无法解决,但可以在并行分布式模式下被解决。
2.3 并行的Cochran-Armitage 算法的设计
1) 串行算法问题的解决并行分布式模式可以解决上述串行算法存在的问题,并且执行效率更高,对资源的消耗更少。
①分布式并行化方法1:将数据的读取分别交由多台机器,一些机器读取aGroup.txt数据,一些机器读取bGroup.txt数据,即可在同一时间开启多个任务并发执行。
②分布式并行化方法2:每台机器都会有自己读取的部分数据,根据计算向数据靠近的原则,在读取数据成功的机器上启动计算逻辑处理数据。这实现了分布式集群计算。
2) 分布式流程
并行分布式整体运行流程如图2所示。首先,主节点提交应用程序主体,为应用程序建立基础运行环境Context。Context负责与集群管理器通信,以及进行资源的申请、任务的分配和监控等。接着,集群管理器为执行器分配资源,启动执行器进程。执行器运行情况会随着“心跳”发送到集群管理器上。然后,Con?text根据程序的依赖关系构建有向无环图(DAG) ,将DGA提交给DGA调度器解析。DAG被切分成多个阶段(stage,每个stage都是一个任务集),Context计算出各个stage之间的依赖关系后将任务集提交给任务调度器进行处理。执行器向Context申请任务后,任务调度器将任务分发给执行器运行。同时,Context将应用程序代码发放给执行器。最后,执行器执行任务,将执行结果反馈给任务调度器和DAG调度器。任务执行完毕后写出数据并释放资源。
图2在逻辑上描述了整个分布式执行流程,包括使用的组件以及各个组件之间的通信与数据传递关系。同时描述了工作节点所需要执行的任务以及任务之间的联系。每个工作节点都有自己的执行器进程,目的是当工作节点执行多个应用程序时,将应用程序彼此隔离。
3 并行分布式Cochran-Armitage 算法的实现
本文将Cochran-Armitage趋势检验算法与大数据分布式计算框架Spark相结合。Spark分布式计算框架可将算法整体分割成数个小任务单独运算,并将每个小任务单独运算的结果整合。Spark借助分布式文件系统HDFS存储数据文件,分布式文件系统HDFS 可将大体量数据文件切割成块(Block) 。这可在处理大数据文件中实现最小化寻址开销。
3.1 分布式集群的基本架构
普通的文件系统只需要单个计算机节点(由处理器、内存、高速缓存和本地磁盘构成)就可以完成文件的存储和处理。分布式文件系统把文件分布存储到多个计算机节点上,成千上万的计算机节点构成计算机集群。目前的分布式文件系统所采用的计算机集群都是由普通硬件构成,这大大降低了硬件上的开销。集群中的计算机节点存放在机架上,每个机架可以存放多个节点,同一机架上的不同节点之间常通过以太网互联,多个不同机架之间采用网络或交换机互联。本文描述的Data Node和Name Node均为机架中的單一节点。分布式集群的基本架构如图3所示。
3.2 分布式存储数据
本文数据都存储在分布式文件系统HDFS中。存储过程如图4。
如图4所示,被Cochran-Armitage 算法处理的基因文件通过HDFS客户端上传,具体分如下4步:① HDFS客户端(HDFS Client) 获取文件信息,将文件切成多个块(Block) 。每一个Block的大小默认是128M。切分完成后,Client保存文件的切片信息,用于下一步的文件上传工作。
② HDFS Client创建Distributed File System对象。Distributed File System对象负责与Name Node节点通信,主要工作有以下两个:工作1是将文件上传请求发送给Name Node,并接受Name Node对文件上传请求的响应;工作2是向Name Node请求获取Data Node,并接受Data Node节点信息。Data Node存储上传文件。
③ 当Name Node响应Data Node的信息后,Client 会与相应的Data Node通信。
④ HDFS Client 收到Data Node 的应答后开始上传数据块。HDFS Client创建FSData Output Stream对象开始文件传输。
3.3 分布式读取数据
执行分布式并行计算所需要的数据均存储在分布式文件系统HDFS。所以在开始计算之前需要读取数据。具体读取过程如图5所示。
① HDFS Client 创建Distributed File System 对象负责与Name Node进行通信。Name Node返回文件块Block所在Data Node的元数据信息。其中包括Block 大小和Data Node信息等。
② HDFS Client创建FSData Input Stream对象,并根据Name Node返回的Data Node信息向Data Node发送读取文件块请求。
③ Data Node接收请求后,开始将文件以字节流的形式发送给HDFS Client。
3.4 分布式并行计算的Map阶段
Map阶段是分布式并行计算的第一步,其核心处理逻辑在map 函数。所有经过图5 中所示的HDFSClient读入的数据都会以键值对形式交给map 函数处理。map函数会对基因数据做转换,并将转换后的数据传输到Reduce 阶段处理。Map 阶段流程如图6。
① Record Reader对象组合Input Format对象,将基因数据块Block以键值对形式从HDFS读入。键值对中的键为数据的偏移量,键值对中的值为读取到的一行数据。
② Map Task(Map 任务)中由map函数处理数据,具体过程如图7所示。
③ Map Task(Map任务)生成的中间数据会根据Hash 分区算法放到对应的bucket(桶)中。每一个Map Task根据Reduce Task(Reduce任务)的数量创建相应的bucket。bucket的数量为m × r 个。其中m 是Map Task的个数,r 是Reduce Task的个数。
④ Bucket中的临时数据会在Reduce阶段被再次抓取和处理。
Map阶段主要依靠map函数处理数据。map函数会对基因数据做转换,并将转换后的数据传输到Re?duce阶段处理。被切分后的基因数据块通过HDFSClient从HDFS读入Spark计算框架。基因数据块中的所有数据需要被Map阶段中map函数处理。基因数据以键值对的形式读入。其中键K为数据在文件中的偏移量,值V为文件中的一行数据。在Spark 以图5方式读取数据块的过程中,对数据做了预处理,删除了K,只保留了V。map函数的具体实现逻辑如图7所示。
① 预处理后数据格式为chromosome-ID:chromosome-start-position:chromoso-me-stop-position;GROUP-NAME|allele1|allele2。其中chromosome-ID 表示染色体号;chromosome-start-position 和chromosome-stop-position 表示基因的起止位置;GROUP-NAME 表示组别编号;allele1 和allele2 分别表示等位基因。
②切割字符串获取等位基因allele1和allele2并更新列联表。Cochran-Armitage算法是对2行3列的列联表做计算。列联表为二维。这里将列联表中每组的数据表示成长度为15的一维表。因为每个等位基因对应一个2行3列的列联表,列联表的行表示分组。本文用到的数据集有5个等位基因,2个组别。所以每个组中一个等位基因要使用3个数据位,5个等位基因使用15个数据位。
③因为更新的一维列联表只能属于一个分组,所以要为列联表标记其属于哪个分组。标记后产生的结果以及中间文件的数据分两部分,一部分表示分组,另一部分表示一维列联表。
3.5 分布式并行计算的Reduce 阶段
Reduce阶段中的数据来自它的上一个Map阶段生成并存储在bucket的中间文件。文件中的数据均由Reduce Task中的reduce函数处理。被处理后的数据会被拉取到主节点进一步处理。具体的Reduce阶段流程如图8所示。
① Map阶段处理后生成的数据文件会有多个分区存储在bucket中。每个bucket对应一个Aggregator。Aggregator 对数据做归并排序操作。Aggregator 本质上是一个HashMap对象。里面的元素为键值对 形式。Aggregator 拉取一个数据,若是HashMap不存在当前key,则插入数据;否则,把value的值累加到V上。
② Reduce Task拉取自己对应分区的数据并交由reduce函数处理。reduce函数处理流程如图9 所示。Reduce Task拉取数据,其中k是分组,v是长度为15的一维表。流程如下:
reduce函数是按照数据的键进行分组,对于键相同的数据每次处理两个。例如:
数据1:
数据2:
对数据1的V与数据2的V在相同索引位相加,得到结果数据::
Reduce阶段完成后,将产生2条键值对数据。一条是
3.6 分布式并行化的趋势检验的实现
趋势检验需要2行3列的二维列联表。因为Re?duce阶段生成的数据是长度为15的一维表,所以需要在逻辑上将一维表切分为5份。每一份代表一个等位基因在对应二维列联表中的一组的数据。图10给出了利用趋势检验算法计算结果并保存的过程:
4 实验结果与分析
4.1 实验数据集
实验所需要的基因数据参考千人基因组计划中的vcf文件[15]生成。本文共使用4组实验数据,每组两个文件。每个文件包含的数据量为:4 946 865条基因数据,12 367 134条基因数据,24 734 269条基因数据和74 202 843条基因数据。每一条均为下列格式的数据:chromosome-ID:chromosome-start-position:chromosomestop-position;GROUP-NAME|allele1|allele2。其中chromosome-ID 表示染色体号;chromosome-startposition和chromosome-stop-position 表示基因在染色體中的起止位置;GROUP-NAME表示分组;allele1和allele2均表示等位基因。chromosome-ID:chromosomestart-position:chromosome-stop-position在计算中用于将同一染色体和同一起止位置的数据分组。GROUP-NAME 用于数据按组别分组。allele1 和al?lele2用于构建列联表。
例:21:9411239:9411239;b|A|A
4.2 实验环境
实验环境分为单机串行和分布式并行。单机串行环境下,计算机系统为Ubuntu18.04,处理器为i7-8750H,内存16G, Java版本为Open JDK 1.8。分布式并行环境下,一共有3台机器,其职责如图2所示。一台机器充当主节点和工作节点的职责,另外两台充当工作节点的职责。3台机器的硬件条件均相同,其计算机系统为Ubuntu18.04,处理器为i7-8750H ,内存16G, Java版本为Open JDK 1.8,Hadoop版本为3.1.3,Spark版本为3.3.0。
4.3 评价指标
本文选择了4个评价指标。(1) 机器数:单机串行环境只有1台机器,分布式并行环境则有大于等于2 台的机器。以不同的机器数量执行任务,所耗费时间一定不同。本文假设执行任务消耗的时间随机器数的增加而减少。(2) 数据量:分布式并行环境应该更能胜任大数据量的情况。数据量越大,串行计算和分布式并行计算的性能差距就越明显。(3) CPU内核使用率和内存利用率:CPU用于执行计算,内存用于交换数据。执行计算机任务应是对资源的利用越合理越好。CPU内核使用越多,则计算效率越高。内存使用越少,资源浪费越少。
4.4 实验结果与分析
① 机器数与任务执行时间的关系
只使用1台机器则属于单机串行环境。机器数大于1台且所有机器执行同一任务则属于分布式并行环境。本文分别使用了1台、2台和3台机器执行趋势检验任务,每一次执行的任务和使用数据集均相同。不同情况下的时间花费如图11所示。相同数据集相同任务情况下,执行时间随着机器数量的增长而减少。这说明并行分布式环境在相同数据集相同任务情况下的执行效率优于单机串行环境。
② 数据量与任务执行时间的关系
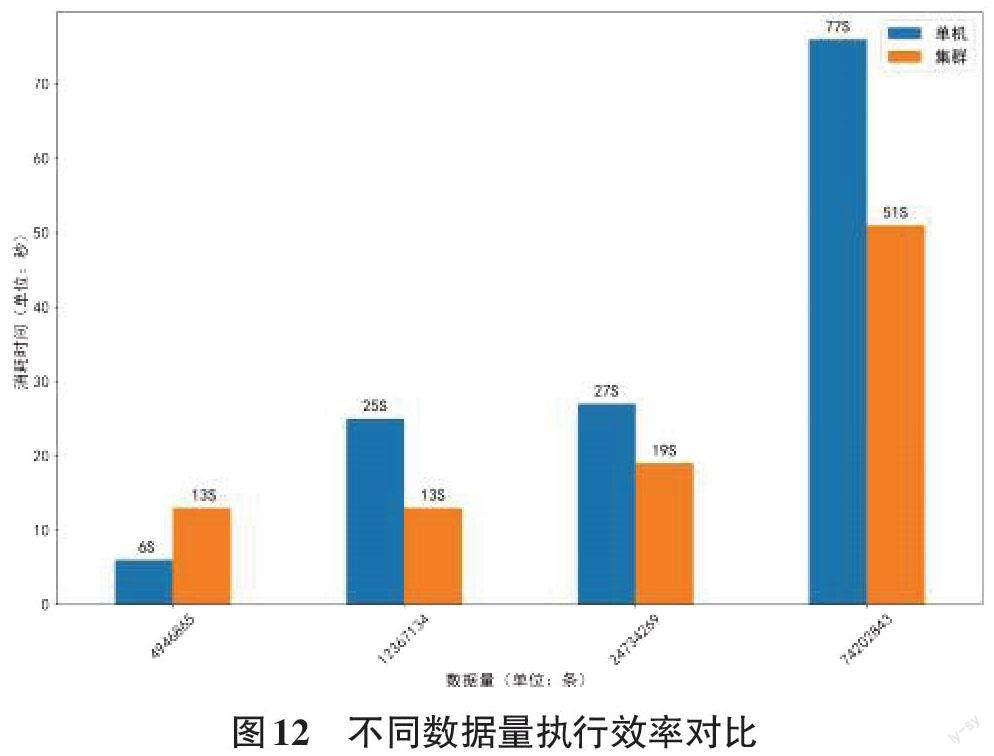
在单机串行环境和三台机器组成的分布式并行集群环境下运行相同任务。使用4组数据,每组数据包括2个基因数据文件。4组数据中每个基因数据文件分别包含4 946 865 条数据、12 367 134 条数据、24 734 269条数据和74 202 843条数据。结果如图12 所示。可知除第一组数据外,分布式并行集群环境下其余组执行任务消耗的时间均小于单机串行环境。因为分布式并行集群启动时需要初始化,所以执行第一组数据的任务时,分布式并行集群环境消耗的时间大于单机串行环境消耗的时间。由图12可说明,分布式并行集群环境与单机串行环境相比更适合大数据量的情况。数据量越多,集群环境所表现的性能越出色。
③ CPU内核使用率
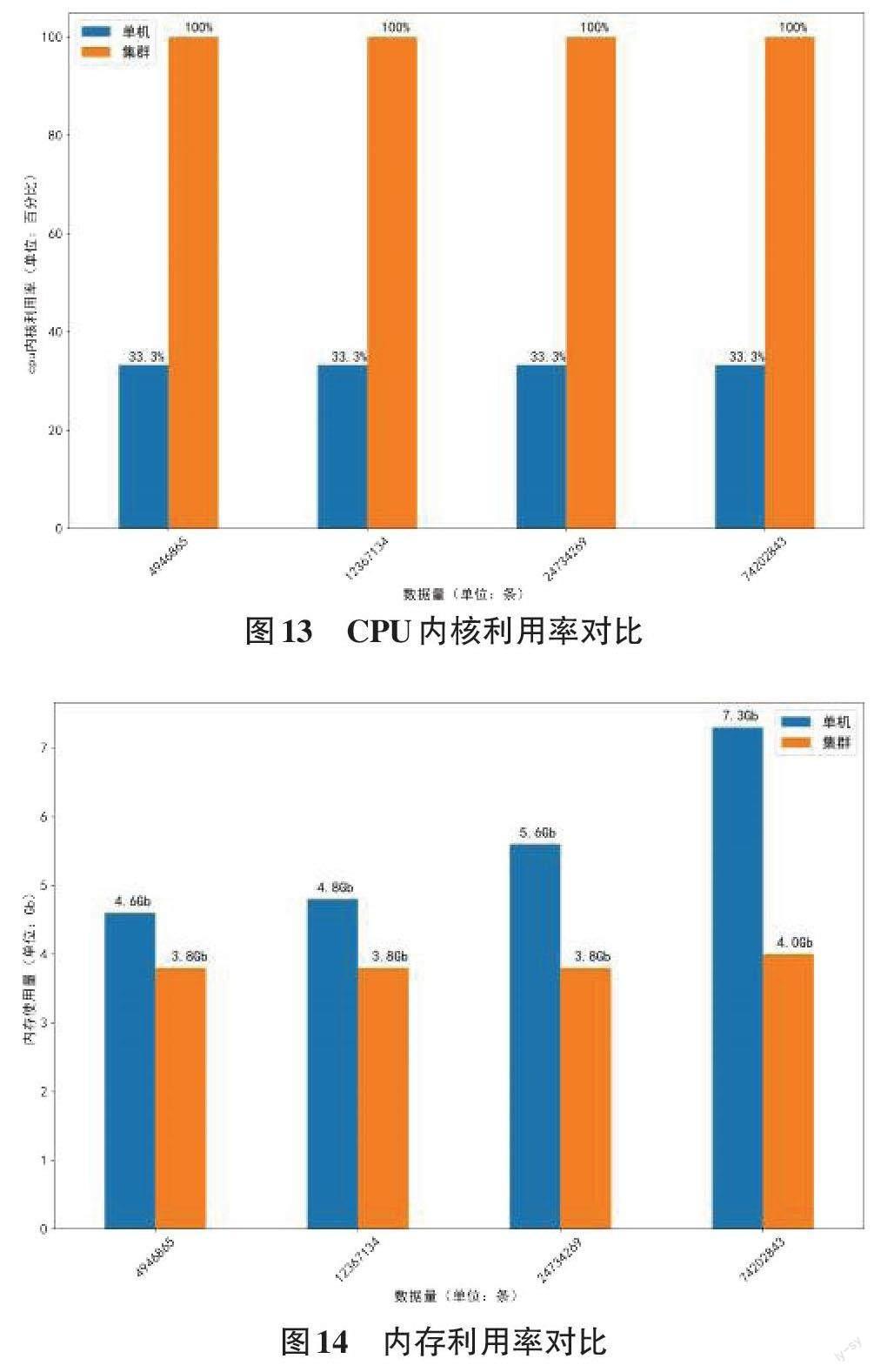
CPU执行计算。其内部有多个核心,每个核心可独立执行任务。对于一次计算任务,使用的核心越多则使用的计算资源越多,计算速度越快。对于此评价指标,本文使用单机串行环境和三台机器组成的分布式并行集群环境运行相同任务。使用4组数据,每组数据包括2个基因数据文件。4组数据中每个基因数据文件分别包含4 946 865 条数据、12 367 134 条数据、24 734 269条数据和74 202 843条数据。结果如图13所示。结果表明,单机串行执行任务总是单线程的,既只使用单个CPU内核。而分布式并行集群执行任务则会使用所有CPU内核。由此表明,分布式并行环境相比于单机串行环境能更有效地使用计算资源,更快的执行任务。
④ 内存利用率
原始基因数据存储在分布式文件系统HDFS中。當分布式并行化Cochran-Armitage算法运行时加载的数据,以及对数据处理产生的中间结果都会放在内存中。内存作为计算机资源的一种,也应该尽量降低利用率,以使内存为更多的程序使用。如图14是在单机串行环境与分布式并行集群环境中对4组数据的内存利用率对比。每组数据包括2个基因数据文件。4组数据中每个基因数据文件分别包含4 946 865条数据、12 367 134条数据、24 734 269条数据和74 202 843条数据。由此表明,单机串行环境对内存的消耗随着数据量的增多而显著增加。分布式并行环境对内存的消耗随着数据量的增加并没有太明显的变化。分布式并行环境对内存的消耗明显的小于单机串行环境。
5 结论
本文设计了分布式并行Cochran-Armitage趋势检验算法的Map阶段的计算过程与Reduce阶段的计算过程,并将任务提交至大数据计算框架Spark,执行Map和Reduce计算过程。本文使用了四组数据集,每组数据包括2个基因数据文件。4组数据中每个基因数据文件分别包含4 946 865条数据、12 367 134条数据、24 734 269条数据和74 202 843条数据。4组数据均使用串行Cocharan-Armitage 算法和分布式并行Cochran-Armitage算法进行了对比。通过上述的4个评价指标可得出结论,分布式并行的Cochran-Armitage 算法明显优于传统单机串行的Cochran-Armitage算法。分布式并行算法相比于串行算法可在很大程度上减少计算时间以及更合理利用和分配计算资源。
【通联编辑:王力】