Recent advances in visible light-mediated chemical transformations of enaminones
2024-04-05YuHnLiyunZhouChengyuWngShngtiFengRongJiePingWn
Yu Hn ,Liyun Zhou ,Chengyu Wng ,Shngti Feng ,Rong M ,Jie-Ping Wn,*
a College of Chemistry and Chemical Engineering,Jiangxi Normal University,Nanchang 330022,China
b School of Chemistry and Chemical Engineering,Linyi University,Linyi 276000,China
c Linyi Sanfeng Chemical Co.,Ltd.,Linyi 276000,China
Keywords: Enaminone Visible light C-H functionalization C=C bond cleavage Cyclization Multicomponent reactions
ABSTRACT Enaminones,which possesses both the nucleophilic enamine as well as electrophilic enone structures,are well known versatile building blocks in organic synthesis.Meanwhile,visible light-mediated reactions have emerged as useful synthetic strategy with enhanced sustainability.Around the last decade,various photochemical transformations of enaminones have been developed to construct cyclic or acyclic compounds.In this review,we describe the recent advances in visible light-mediated chemical transformations of enaminones.Detailed discussion on the reaction mechanism of the related reactions is given to provide guide to the reader.Finally,a summary on the existing challenges and the future outlook towards the development of practical photocatalytic reactions of enaminones is also presented.
1.Introduction
Enaminones or enaminoesters,which consist of an amino group linked through a carbon-carbon double bond to a carbonyl or ester group,are important and powerful synthons [1-7].Owing to the amine-alkene-carbonyl conjugated structural features,they combine the triple nucleophilicity (C-2,amino and carbonyl oxygen)with the ambident electrophilicity of enones (C-1 and C-3) (Fig.1).Enaminones have been utilized as versatile building blocks in organic the synthesis of carbocyclic [8-11],heterocyclic [12-18] as well as acyclic compounds [19-23] under proper reaction conditions.The known transformations usually take place in the unit of“enamines” or “enones” in the presence of one or more other reaction partners,enabling the construction of heterocycles though tandem cyclization under proper environments [24-27],the functionalization of the internally activated vinyl C-H bond under the catalyst of transition metal [28-30,27],iodide [31-34] or other oxidants [35,36],C-H bond functionalization of aryl ring in the aromatic ketone or aryl amine moiety [37-39],the synthesis of acyclic compounds through the cleavage of the C=C double bond by means of different pathways of transformation [40-43],the functionalization of the C-N bond to different enones [44,20],as well as some other novel reactions in enaminones [45-49].The results,especially the ones reported over the last decade by us and others,have opened new frontiers in the enaminone-based synthetic chemistry.

Fig.1.Structure of enaminones.
Following the daily increasing emphasis on sustainable synthesis,the visible light-mediated reactions has won significant attention and advances [50-52] since the pioneer reports by MacMillan [53-55],Yoon [56,57],Stephenson [58-60] and Nicewicz [61-63].By employing visible light photocatalysis,a large variety of new synthetic reactions have been established to deliver diverse organic productsviamild,easy to handle,and environmentally benign operations.Such reactions are more favorable than conventional heating protocols from the perspective of green chemistry.In this regard,applying photocatalysis to the chemical transformations of enaminones could be valuable approaches for organic synthesis.Actually,during the last decade,a broad range of synthetic works on the functionalization of enaminones under visible light catalysis have been published.However,no review work on the photocatalytic reactions of enaminones is currently available.Thus,we present herein an overview on the progress of visible light-mediated chemical transformations of enaminones,in hope of bring some guides to the development of more environmentally friendly synthesis by the transformation of enaminones.
2.Direct α-C(sp2)-H bond functionalization
In recent years,the direct functionalization of the alkenylα-C(sp2)-H bond in enaminones has gained extensive attention because of its highly versatile reactivity and widespread application in designing the synthesis of biologically active molecules and pharmaceutical substances.Among these approaches,visible lightmediated directα-C(sp2)-H bond functionalization is definitely one of the simple and ideal methods.Recently,several directαfunctionalization reactions of enaminones such asα-selenylation,α-fluoroalkylation andα-thiocyanation have been developed.
2.1. Photocatalytic α-C-H selenylation of enaminones
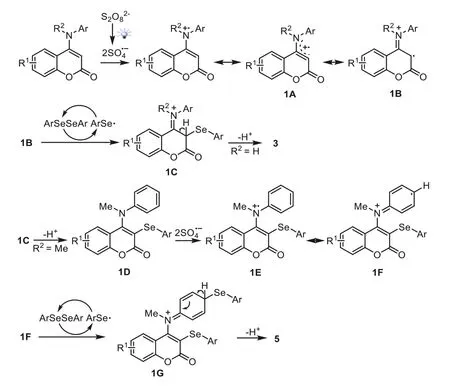
In 2018,Yang and coworkers reported the direct C(sp2)-Hα-selenylation of coumarin derivatives under metal-and photocatalyst-free visible light catalysis [64].The reactions were run with the conditions of (NH4)2S2O8as the oxidant,CH3CN as the solvent under air atmosphere as well as the irradiation of 12 W blue LEDs visible light at room temperature.As shown in Scheme 1,aryl selenium could be smoothly incorporated to various coumarin derivatives atα-site.A wide array of diselenides and coumarin derivatives,bearing either electron-withdrawing or electron-donating groups,were tolerated in this reaction,leading to the desired product in generally good to excellent yields.It is worth noting that as toN-substituted 4-(phenylamino)-2Hchromen-2-ones4,the dual selenylated products were selectively generated in 71%-81% yields (Scheme 2).When thepara-site of the 4-phenylamino group was substituted,only C-3 selenylated products were obtained.Two possible mechanisms to rationalize the formation of3and5were illustrated (Scheme 3).First,theinsitugenerated active radical anion SO4·-from (NH4)2S2O8occurred under the irradiation of visible light.Then,a single electron transfer (SET) process between1and SO4·-to delivered the radical cation1Aor1B,which further reacted with diselenides2to form the intermediate1C.Finally,if R2was H atom,the final product3was obtained by the elimination of H+from1C.If R2was methyl,the reaction of intermediate1Cwith diselenides2led to the C-3 selenylated species1D.Subsequently,a SET process took place between1Dwith SO4·-to give radical cation1Eor1F,which was then captured by ArSe· to afford the intermediate1G.The deprotonation of1Gafforded the final product5.

Scheme 1.Visible-light-promoted regioselective selenylation of 4-(phenylamino)coumarins.

Scheme 2.Visible-light-induced selenylation of N-substituted 4-(phenylamino)coumarins.
Scheme 3.Proposed mechanisms for the formation of 3 and 5.
2.2. Photocatalytic α-C-H perfluoroalkylation of enaminones
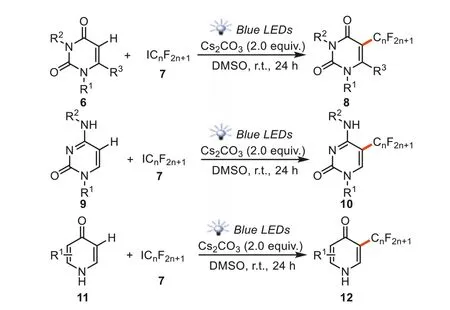
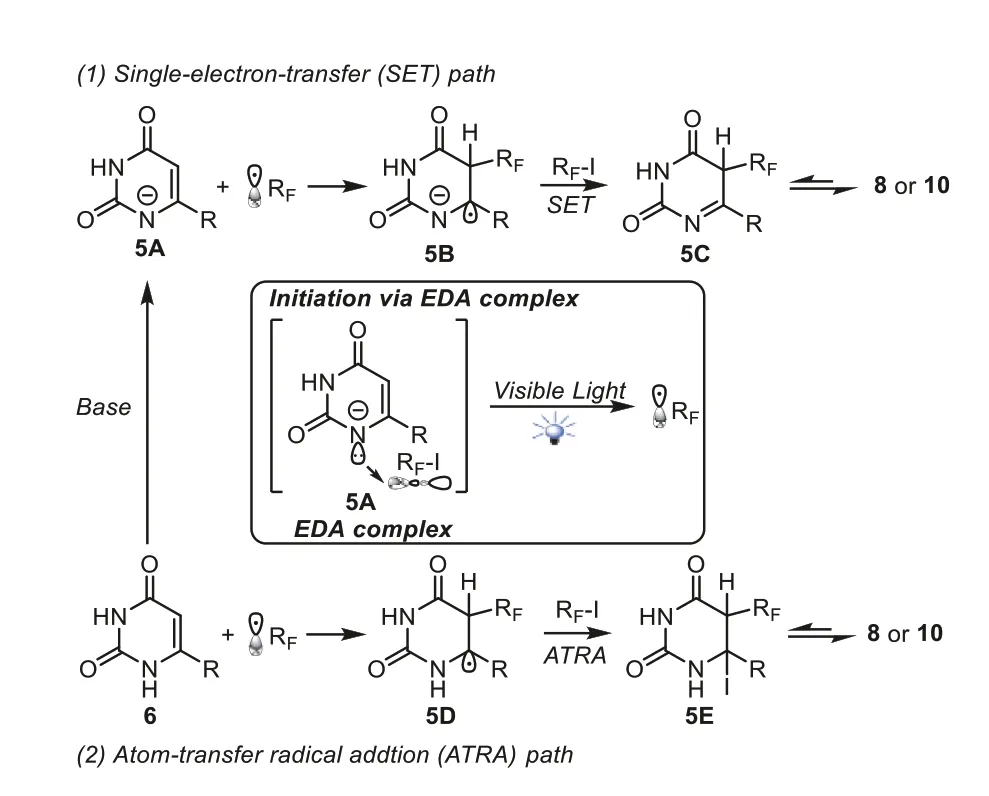
Incorporation of fluoroalkyl groups into a molecule is important because such elaboration can dramatically alter the chemical and physical properties of the molecules and results in various applications in pharmaceuticals,agrochemicals,and functional materials[65].In 2018,Heetal.reported catalyst-free and visible light promoted trifluoromethylation and perfluoroalkylation of uracils,cytosines and pyridiones in theα-C-H bond of the enaminone fragments (Scheme 4) [66].The reactions were conducted with 12 W blue LED irradiation at room temperature using Cs2CO3as base and DMSO as solvent.The negatively ionized uracil5Awas first generated in the presence of Cs2CO3.A fluoroalkyl radical could be formed under the irradiation of blue LEDsviathe EDA complex.Then two pathways might be involved in the propagation step: (1) fluoroalkyl radical reacted with5Ato produce intermediate5B,which accessed to intermediate5Cviaa SET process in the influence of fluoroalkyl iodides.Finally,the desired product8or10were formed by 1,3-hydrogen migration process with the help of Cs2CO3.(2) The Rfradical might add to uracils6and generate the carbon radical intermediate5D,which abstracted an iodine atom from RfI to afford5E.The final products8or10were obtainedviathe fast re-aromatization of5EviaHI elimination (Scheme 5).
Scheme 4.The synthesis of α-perfluoroalkylated uracils,cytosines and pyridiones.
Scheme 5.Proposed mechanisms for the formation of 8 and 10.
2.3. Photocatalytic α-C-H thiocyanation of enaminones
Organic thiocyanates are moieties possessing plentiful biological and pharmaceutical activities in both synthesized and naturally occurring molecules [67].Among the reported strategies on organic thiocyanate synthesis,the direct C-H thiocyanation is more practical and cost-effective because such a method allows for the synthesis of organic thiocyanates using those prevalently available C-H bond donors.In 2019,our group realized the first vinyl C-H bond thiocyanation reaction of tertiary enaminones under metal-free,photocatalytic conditions in the presence of Rose Bengal,which enables the synthesis of thiocyanated alkene derivatives and chromones using NH4SCN as the thiocyano source under an aerobic atmosphere [68].Besides,employing Ru(bpy)3Cl2·H2O as the photocatalyst switches the reaction pathway to provide NH2-functionalized thiocyanated enamines.As shown in Scheme 6,the reactions might start from the quenching of NH4SCN to the excited RB*species generated from the visible light irradiation to the RB catalyst,which gave rise to the SCN free radical.Meanwhile,the RB·-formed therein could be regenerated to RB by the oxidation of molecular oxygen.Subsequently,the addition of the SCN· to the C=C double bond in13provided the free radical intermediate6A,which was further oxidized to carbon cation6B.The deprotonation on6Bthen yielded thiocyanated product14.when the Ru-catalyst is employed,a transamination between the tertiary enaminones and the ammonium was promoted to form products15,wherein the Ru-catalyst might act also as a Lewis acid (LA) catalyst.In addition,wheno-hydroxylphenyl-functionalized enaminones were used as substrates,the successive chromone annulation took place to give thiocyanated chromones16.

Scheme 6.Photocatalytic synthesis of polyfunctionalized alkenes and thiocyano chromones.
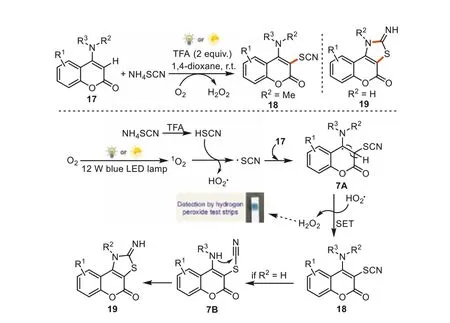
After our work,Yangetal.reported the photocatalyst-free C-H thiocyanation of 4-anilinocoumarins by visible light irradiation(Scheme 7) [69].The reactions started from ammonium incorporation of thiocyanate and TFA forming thiocyanic acid (HSCN).Under the visible light irradiation,oxygen was converted to singlet oxygen which subsequently abstracting an electron from HSCN to generate HO2· and ·SCN.The selective addition of thiocyano free radical to the coumarin derivatives17gave the intermediate7A.The SET processes from HO2· to the intermediate7Adonated the desired products18by releasing H2O2.If R2was H,the intramolecular nucleophilic addition of NH to CN afforded the cyclization products19.
Scheme 7.Photocatalyst-free regioselective C-H thiocyanation of 4-anilinocoumarins.
Despite the fact thatN,N-disubstituted andN-mono substituted enaminones have been successfully used as the C-H bond donors for C-H thiocyanation by photocatalysis,equivalent photocatalytic C-H thiocyanation reactions of the free NH2-enaminones kept inapplicable in the aforementioned systems.In 2022,our group reported the firstα-C-H thiocyanation of NH2-enaminones with air oxygen as the oxidantviavisible light photocatalysis [70].By using NH2-enaminones20and NH4SCN as starting materials,the synthesis ofα-thiocyanated NH2-enaminones21could be accessed by means of blue LEDs irradiation in the presence of Ru(bpy)3Cl2·6H2O photocatalyst.The reactions were initiated by the Ru-based photocatalyst enabling the formation of ·SCN and PC anionic radical.The free radical addition of ·SCN to the enaminone C=C double bond in20donated8A.Meanwhile,the coupling of the PC anionic free radical to O2leads to the generation of an oxygen anionic free radical accompanied by PC regeneration.Intermediate8Acaptured oxygen anionic free radical (observed by HRMS) afforded intermediate8Bin the presence of an ammonium cation.Further,the elimination of hydrogen peroxide in intermediate8Bgenerated thiocyanated NH2-enaminones21.When base was used following the photocatalytic process,the tandem cyclization and tautomerization processes took place to give 2-aminothiazoles22.On the other hand,when NH2-enaminones21are employed in aqueous acidic conditions,2-thiazolinones23were obtainedviacyclization,hydrolysis,and the elimination of ammonium (Scheme 8).
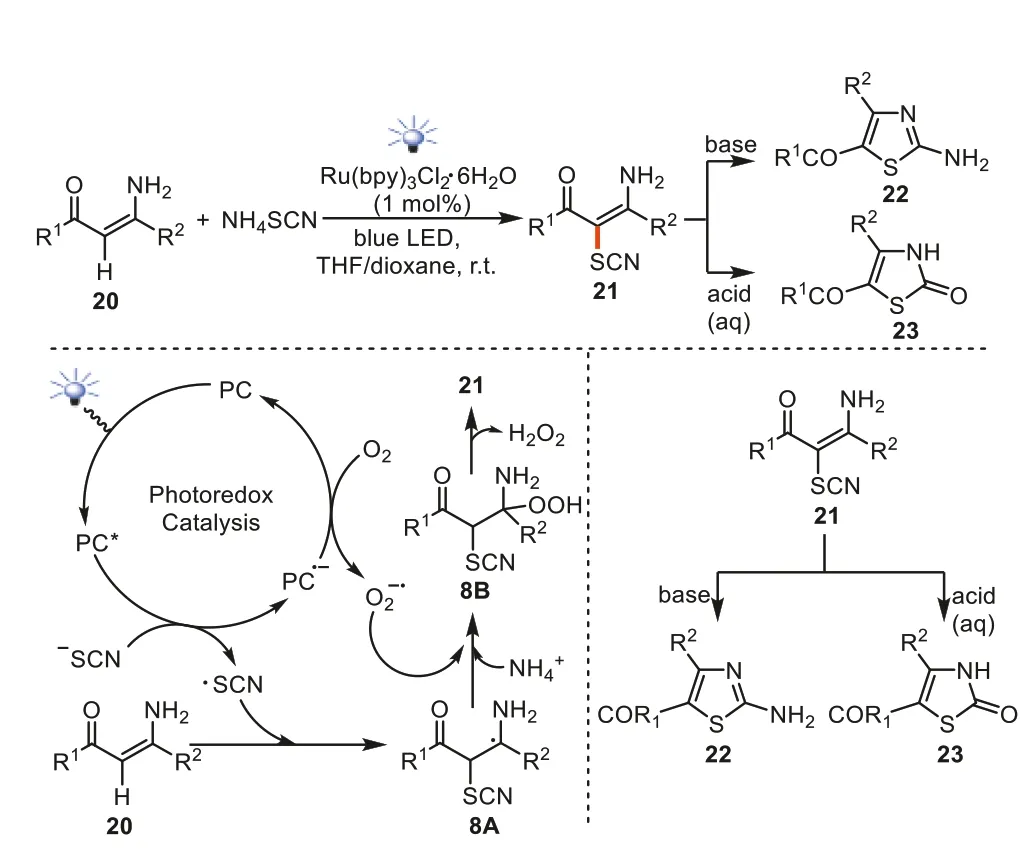
Scheme 8.Photocatalytic C-H thiocyanation of NH2-enaminones.
3.Functionalization reactions via C=C double bond cleavage
Scissoring the C=C bond of enaminones constitutes one of the irreplaceable tools of modern organic synthesis in the form of either partial [71-74] or full bond cleavage [75-77].Among them,visible-light-mediated C=C bond cleavage of enaminones have been also identified as useful synthetic approaches.
3.1. Synthesis of 1,2-diketone or its derivatives
As early as in 1970s,Wasserman and Ives firstly revealed that 1,2-diketones can be obtained from enaminonesviaC=C double bond cleavage under the conditions of 650 W lamp heating at -78°C to room temperature [78,79].In these reactions,the C=C bonds of enamines were cleaved through [2+2] cycloaddition procedure to give two carbonyl compounds.In 2015,our group developed a facile method for the synthesis of 1,2-diketonesviaC=C bond cleavage of enaminones under ambient conditions through visiblelight photocatalysis in presence of Rose Bengal (RB) [80].By employing enaminones24as the substrate,as shown in Scheme 9,with visible light irradiation,RB was converted into the excited RB*,which further reacted with3O2to regenerate the stage of RB and produce singlet oxygen1O2.The singlet oxygen then incorporated to the enaminone24,giving peroxide intermediates9A.The subsequent decomposition of9Agave the 1,2-diketones25,which could be captured by diamine to afford quinoxalines26viastepwise operation.

Scheme 9.Synthesis of 1,2-diketones and quinoxalines via C=C bond cleavage of enaminones.
Interestingly,by employing enaminones27to react with alcohols28under the irritation of 20 W green LEDs in the presence of Rose Bengal,our group further developed a facile method for the synthesis of usefulα-ketoesters [81].This method showed general tolerance to the aryl group of enaminones and primary/secondary alcohols.Analogously,the visible light irradiation to RB gave rise to excited RB*species,which further activates3O2to the active singlet oxygen1O2.The highly active singlet oxygen1O2then coupled the C=C double bond of enaminone to offer 1,2-dioxetane10Awhich was further converted to intermediate10Bthrough the ring opening and a subsequent N-O bond formation process.Next,alcohol acted as nucleophilic reagent to attack the C-O bond in intermediate10B,delivering zwitterion intermediate10C.The successive decomposition afforded the final productα-ketoester29and10D.The hemiaminal10Dmight be further oxidized to DMF under the titled oxidative conditions (Scheme 10).
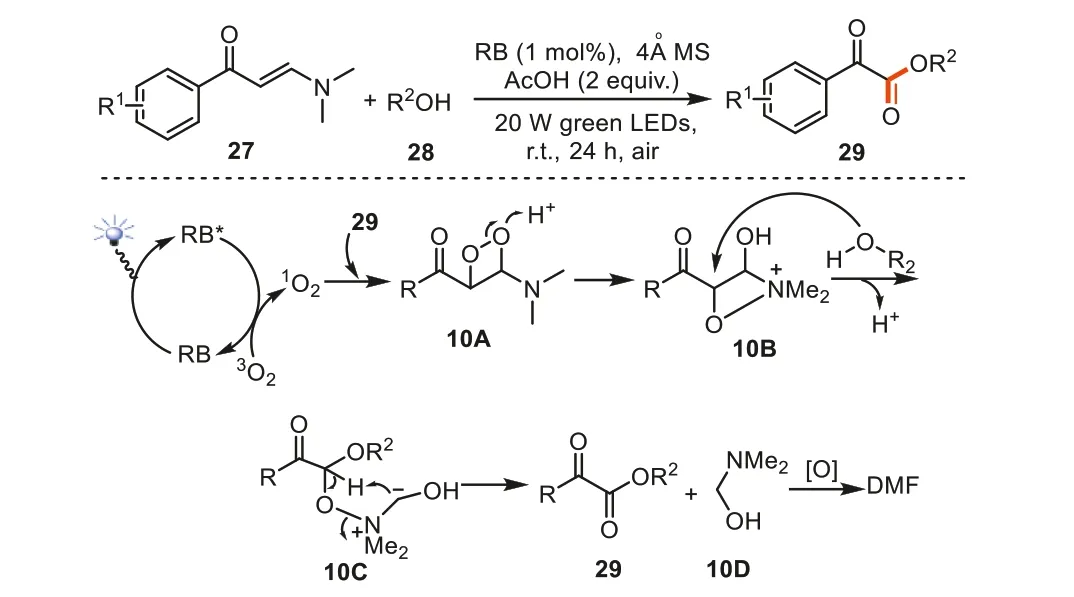
Scheme 10.Synthesis of α-ketoester via C=C bond cleavage of enaminones.
3.2. Synthesis of quaternary amino acid derivatives
In 2014,Li and co-workers reported a visible-light-promoted transformation involving the ene-type reaction of secondary enamino ketones with singlet oxygen,followed by a 1,2-acyl migration,affording quaternary amino acid derivatives [82].The reaction pathway was distinctively different from the previous reports of C=C bond cleavage by singlet oxygen.As shown in Scheme 11,under the irradiation of 14 W CFL,ruthenium(II) is converted into a high-energy excited singlet1RuII*,which underwent intersystem crossing (ISC) to triplet3RuII*.Then the triplet3RuII*species reacted with3O2viaintermolecular energy transfer process to regenerate the ground-state RuIIand produced the reactive1O2species.Next,an ene-type reaction happened between30and1O2to yield the intermediate11A.The dehydration of11Agave11B,and a nucleophilic addition of alcohol to11Bled to11C.With subsequent 1,2-acyl migration and protonation,the products31were formed (Scheme 11).It should be noted that a fluorescence emission quenching study and the redox potential of the substrate30measured by cyclic voltammetry experiments were carried out to support the proposed mechanism.
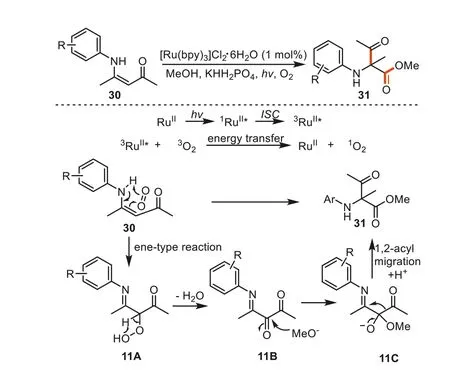
Scheme 11.Visible-light-mediated the partial cleavage of enaminone C=C double bond.
3.3. Synthesis of α-substituted γ-ketoesters
In 2022,Jin and co-workers developed a blue visible-lightpromoted approach for the synthesis ofα-substitutedγ-ketoester derivativesviaC=C bond partial cleavage of enaminones by using enaminones32and diazoesters33as the substrate [83].The blue visible light irradiation induced the selective photolysis of aryl diazoesters33to give free carbene species with by releasing nitrogen gas.Theinsituformed free carbene species was captured by enaminones32to provide the cyclopropane intermediate12B.Due to the stronger electron-withdrawing effect of arylcarbonyl group than that of ester carbonyl group,path a instead of path b could occurviaring opening driven by nitrogen atom,affording the intermediate12Cwhich might be further converted intoα-formyl arylγ-ketoeser34viahydrolysis.Then,with the help of Al2O3Lewis acid,the final productα-substitutedγ-ketoester35could be obtained by decarbonylation of34(Scheme 12).This strategy broadened the methods available for accessing theα-formyl arylγ-ketoester orα-arylγ-ketoester derivatives.

Scheme 12.Synthesis of α-substituted γ-ketoesters via C=C bond cleavage of enaminones.
4.Annulation reactions for the synthesis of heterocyclic and carbocyclic scaffolds
4.1. Synthesis of five-membered N-heterocycles
As a typical five-memberedN-heterocycles,the indole motif is a well-documented core structure of numerous natural products and bioactive compounds.In addition to the Fischer indole syntheses,many different synthetic approaches using modern transitionmetal catalysis are known.The enaminone annulations leading to indolesviaintramolecular C-H activation by transition metal catalysis have been known for long period,but the reliance on stoichiometric chemical oxidant and heating at elevated temperature are limits of such methods.In 2014,Ruepingetal.reported the enaminone-based indole synthesis using a new combination of palladium and photoredox catalysis [84].With catalytic amount of the photoredox catalyst in the presence of visible light,the typical high loadings of external oxidants could be avoided.In the first step,the C-H bond activation of the olefin of36took placeviaPd(II) insertion,giving the intermediate13Awhich further underwent the arene C-H bond to form the six-membered cyclopalladium intermediate13B.After reductive elimination,indoles37were produced and the resulting Pd(0) was re-oxidized to Pd(II)by either the photoredox catalyst or theinsituformed superoxide anion (Scheme 13).

Scheme 13.Synthesis of indoles via intramolecular annulation reactions of enamine ester.
Very recently,Baell and Huangetal.established a protocol for the synthesis of polysubstituted oxazoles through photocatalytic benzylic C-H oxidation/cyclization of enaminones with molecular oxygen as an ideal and green oxidant [85].This strategy featured broad substrate scope and good functional group tolerance.Based on the results of control experiments and the DFT computations,the authors proposed a plausible mechanism (Scheme 14).Firstly,the Ru(bpy)2(PF6)2was excited by 495 nm LEDs to form Ru(bpy)2(PF6)2*which underwent a SET with the benzylic C-H bond of B-O complex14Ato give Ru(bpy)2(PF6)2·-and benzyl radical14B.The Ru(bpy)2(PF6)2·-is subsequently oxidized by molecular oxygen to regenerate Ru(bpy)2(PF6)2and release superoxide radical anion O2·-.The superoxide anion coupled proton to generate ·OOH.The benzyl radical14Bcoupled with ·OOH to access adduct14Cwhich could be further transferred into oxygen radical14Dby losing PhBpin and hydroxyl radical.Then,two possible reaction pathways could occur.On the one hand,the 2,3-dihydrooxazole skeleton14Gwas formedviaintramolecular cyclization of oxygen radical intermediate14Dand a deprotonation process (path a,Scheme 14).On the other hand,diradical species14Fcould be obtainedviadeprotonation,and the subsequent intramolecular radical-radical coupling gave14G(path b,Scheme 14).Finally,the desired oxazole product39was afforded through further oxidation and deprotonation process under O2.Due to the high electro-negativity of oxygen in the hydroxyl radical,it is relatively unstable and tends to react with the C-H bond to form a more stable carbon radical,path a was hypothesized as the more favorable route.
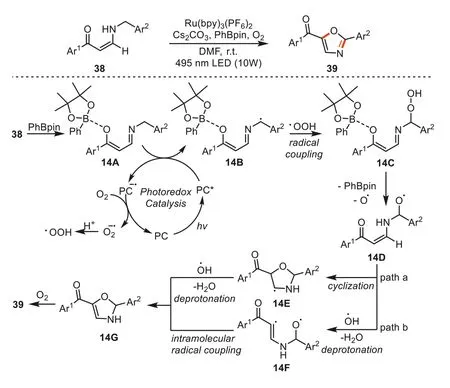
Scheme 14.Synthesis of polysubstituted oxazoles from enaminones.
4.2. Synthesis of six-membered N-heterocycles and O-heterocycles
In 2018,Yu and co-workers achieved the synthesis of quinoxalinesviatandem azidation/intramolecular C-H amination under visible light irradiation by using 1-azidyl-1,2-benziodoxole as the azidating agent withN-arylenamines [86].Under the irradiation of visible light,the I-N bond of15Acleaved to yield the azidyl radical15B,which subsequently added to the C=C double bond inN-arylenamines to form the intermediate15C.The15Cwas further oxidized to carbocation intermediate15D,which was then converted into vinyl azide15Eviaoxidative deprotonation.Vinyl azide15Ewas more prone to be oxidized to imine radical15Gbecause the azide group would render the C=C double bond in15Eelectronically richer than the one in40.Subsequent cyclization of15Gfollowed by oxidation and deprotonation afforded the final products41(Scheme 15).It was worth mentioning that the substituent atβ-site inN-arylenamines40exhibited crucial influence on the reaction,and the conditions were needed to be modified for the reactions of different substrates.For example,Cu(OAc)2was required for the preparation of 3-(trifluoromethyl)quinoxalines,whereas Ru(bpy)3Cl2along with Cu(OAc)2combination was found to be more favorable for the synthesis of 2,3-diarylquinoxalines(Scheme 15).
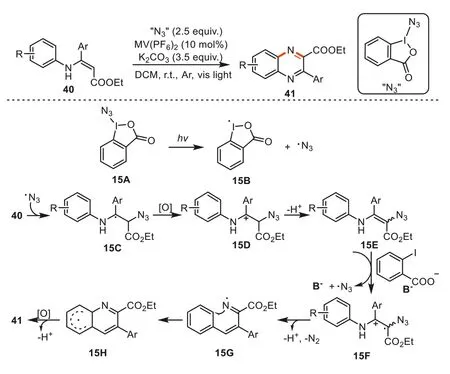
Scheme 15.Synthesis of quinoxalines via tandem azidation/cyclization of Narylenamines.
Currently,the annulation reactions ofN,N-disubstituted 2-hydorxyphenyl enaminones has been widely used in the construction of diversely functionalized chromones,especially for the synthesis of 3-substituted chromones.Among these methods,photoredox catalysis has won particular attention as options of the enhanced sustainability.In 2017,Yang and Chen reported a visiblelight-driven,radical-triggered tandem cyclization ofo-hydroxyaryl enaminones for the synthesis of 3-CF2/CF3-chromones [87].At first,the irradiation of the catalyst [Ir(ppy)3] under visible light generated the excited state Ir(ppy)3*which was then oxidized by BrCF2COOEt or Ph2SCF3OTf to generate [Ir(IV)(ppy)3]+complex and RFfree radical species.Subsequently,the RFradical added to the C=C double bonds of substrate42regioselectively to give radical intermediate16A.Further oxidation to this intermediate by [Ir(IV)(ppy)3]+provided16Bwith the concurrent regeneration of [Ir(ppy)3].Next,the hydroxyl group on benzene ring attacked the iminium cation to give intermediate16C.Ultimately,theN,Ndimethyl group was eliminated to furnish the desired product45/46(Scheme 16).Ten 3-CF2COOEt and ten 3-CF3substituted chromones were obtained in acetone at room temperature in good to excellent yields.
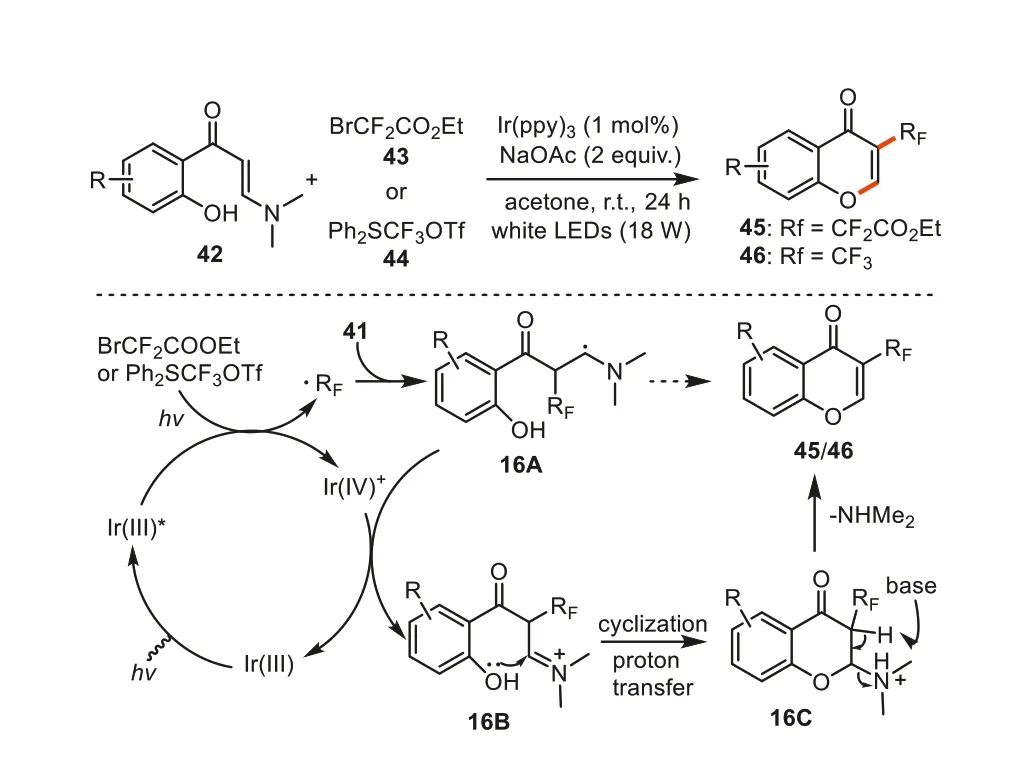
Scheme 16.Synthesis of 3-CF2/CF3-containing chromones via tandem cyclization of o-hydroxyaryl enaminones.
In the same year,Zhangetal.developed an efficient method for the synthesis of 3-CF2-chromonesviaa ruthenium reduction cycle from the similar substrates [88].The catalyst [Ru(bpy)3]2+was firstly converted into the excited state [Ru(bpy)3]2+*under visible light,and enabled the oxidation of Et3N to generate the [Ru(bpy)3]1+species.Then the electron-rich metal complex[Ru(bpy)3]1+underwent a SET process with the cleavage of C-Br bond in CF2BrCOOEt to afford an electron-deficient radical17Aand regenerate [Ru(bpy)3]2+.Intermediate17Aadded to the C=C double bond in substrate42to give radical17B.The SET between17Band [Ru(bpy)3]2+*provided iminium cation intermediate17Cand the metal complex [Ru(bpy)3]1+.Finally,the chromones47were obtainedviaintramolecular annulation and a subsequent elimination of dimethylamine (Scheme 17).
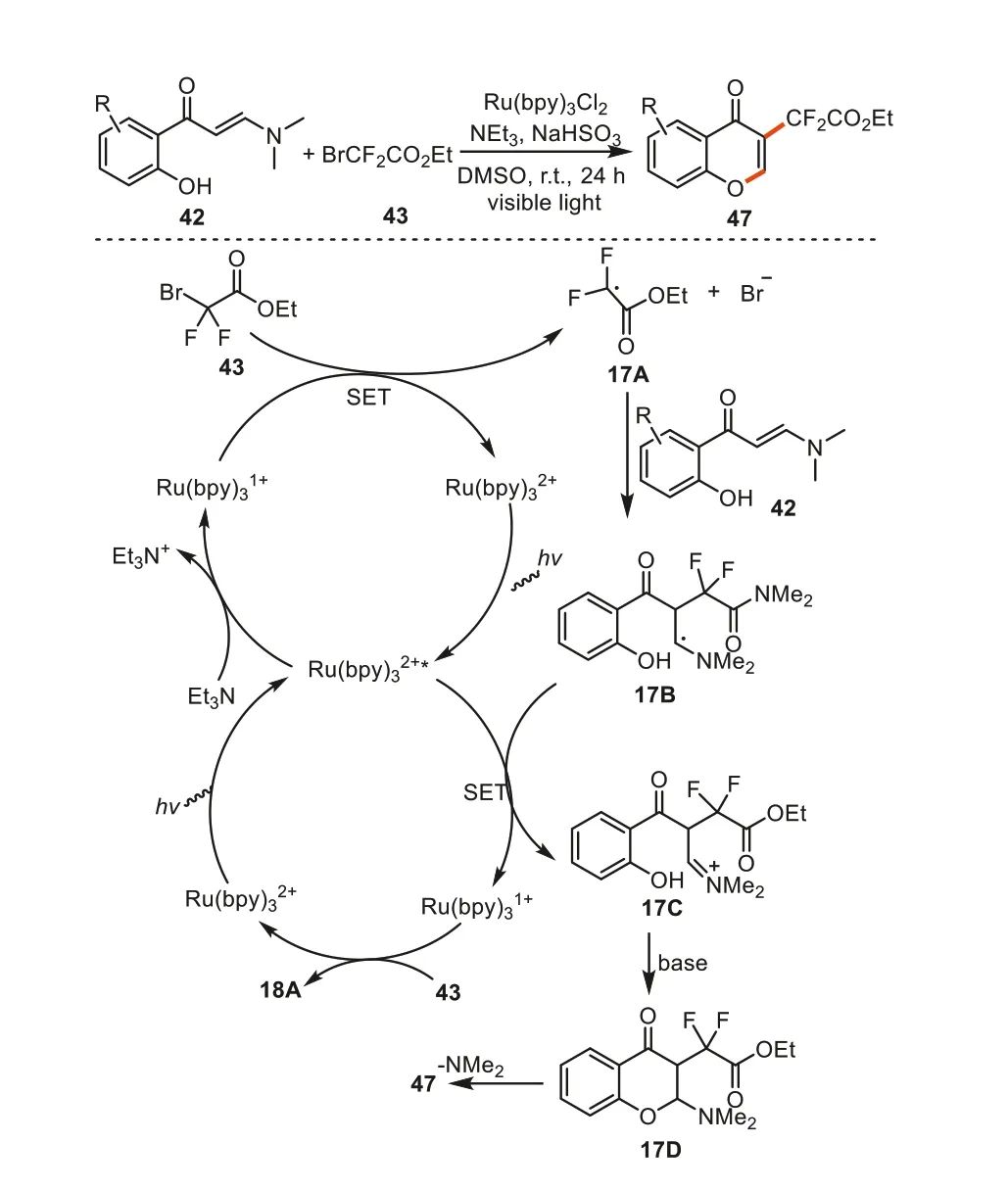
Scheme 17.Synthesis of 3-CF2-containing chromones via radical cascade reaction of o-hydroxyaryl enaminones.
In 2020,Iaroshenko and Mkrtchyan developed two facile routes to synthesis diverse 3-arylchromones through photo-arylation reactions ofortho-hydroxyarylenaminones by using aryldiazonium tetrafluoroborates and diaryliodonium triflates as aryl sources,respectively [89].Initially,the irradiation of Eosin Y and Ru(II) generates their excited states by an appropriate light source.Aryl radicals18Acould be formed from aryldiazonium tetrafluoroborates and diaryliodonium triflates catalyzed by the corresponding excited*Eosin Y and*Ru(II) species.The aryl radicals18Aformed thereby attacked promptly on the enaminone to afford the radical intermediate18Bwhich was oxidized afterwards to carbocation18C.The annulation on this intermediate then gave intermediate18D.Finally,the 3-arylchromones49were furnished by Et2NH elimination (Scheme 18).

Scheme 18.Visible-light-mediated arylation of o-hydroxyaryl enaminones by BF4N2Ar and TfOIAr2.
In 2021,the same group disclosed different methods for the synthesis of 3-arylchromones by arylation oforthohydroxyarylenaminones by photocatalysis by employing sulfonium salts and arenesulfonyl chlorides as the aryl radical precursors[90].These two routes showed good efficiency and were feasible for the preparation of 3-arylchromones in good to excellent yields.Analogously,the pathways were commenced by a SET oxidation initiated by the excited state Ru(II)*,which coupled with triarylsulfonium and arenesulfonyl chlorides to afford aryl radical (Ar·).The addition of the free radical to the enaminone moiety gave carbon-centered radical19Bwhich could be further be converted into the final products49vianucleophilic annulation and amine elimination (Scheme 19).

Scheme 19.Visible-light-mediated arylation of o-hydroxyl aryl enaminones by sulfonium salts and arenesulfonyl chlorides.
In 2021,Xuetal.developed a visible light-promoted,metal-free selenylation/cyclization cascade betweenortho-hydroxyaryl enaminones and diselenides for the synthesis of 3-selanylchromones under mild conditions [91].Notably,no transition metal catalyst,photocatalyst or additional oxidant were required in the reactions.The final products could be easily converted into selenyl-functionalized pyrimidines by reacting with benzamidines.As for mechanism,the light irradiation to diphenyl diselenide53produced phenylselenyl radicals which could be further oxidized to phenylselenyl cation20Aby air.Subsequently,the phenylselenyl cation20Acoupled the C=C bond in the enaminone42to afford cycloselenium ion20Bon which a ring-opening driven by lone pair electrons of NMe2took place to give rise to imine cation intermediate20C.Then the intramolecular cyclization of20Cand a following elimination of dimethylamine delivered the final products54(Scheme 20).
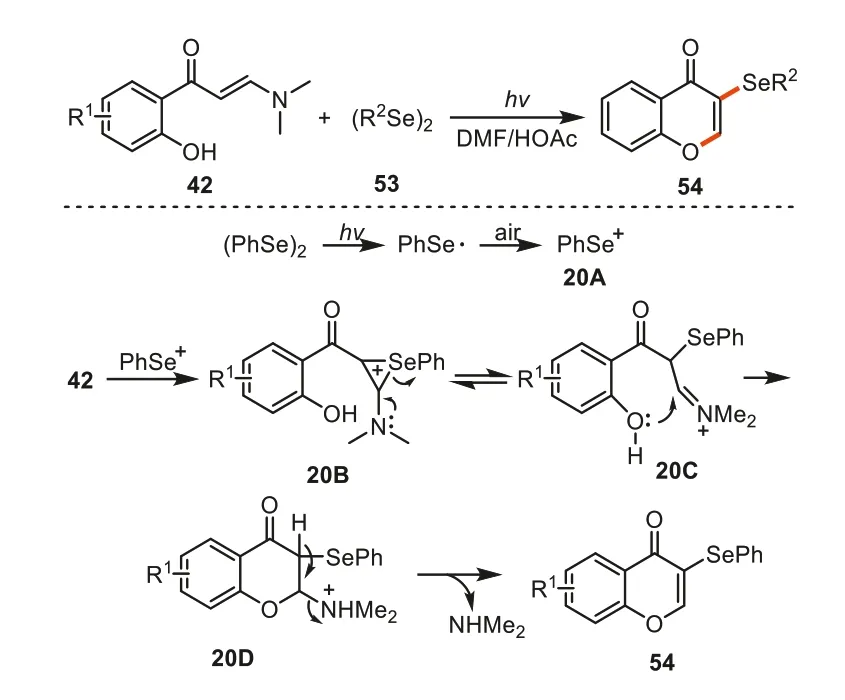
Scheme 20.The photocatalytic synthesis of 3-selenochomones.
In 2022,Leetal.disclosed a method for the synthesis of 3-aminoalkyl chromones fromo-hydroxyphenyl enaminone andNaryl glycines55under visible light catalysis [92].This synthetic method required no photocatalyst or additive,and proceeded to products with good functional group tolerance.The reactions were proposed to be initiated by the light irradiation to enaminones42,affording the excited state21A(42*),which acted as the photocatalyst to provide singlet oxygen (1O2) from molecular oxygen through energy transfer process.The singlet oxygen (1O2) interacted with ammonium-carboxylate salt21B,an isomeric form ofN-arylglycine55,to give theα-amino radical intermediate21Cand superoxide radicals (O2-·).Intermediate21Cwas further oxidized and deprotonated to form the imine specie21Dwhich was subsequently captured by substrate42vianucleophilic addition to provide iminium intermediate21E.The intramolecular cyclization of21Eby intramolecular nucleophilic addition of OH group to imine cation gives intermediate21F.The elimination of dimethylamine then led to the desired products56(Scheme 21).
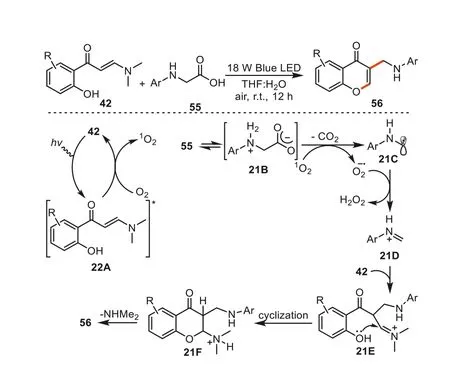
Scheme 21.The photocatalytic synthesis of 3-aminoalkyl chromones.
In the same year,the group reported another approach for the synthesis of 3-aminoalkyl chromones56usingN-arylglycine esters as the alkylating agents by the cooperative catalysis of visible light and an enzyme.By utilizing Methylene Blue (MB) as the photocatalyst [93].This approach avoided the synthesis of acids from esters in a separate step,sinceN-aryl glycines were derived fromN-aryl glycine salts under strong basic conditions.As shown in Scheme 22,Firstly,N-aryl glycine ester57was hydrolyzed intoN-arylglycine22Awith the help of Lipase B Candida antarctica (CALB).Then radical cation22Bcould be formed fromN-aryl glycine22AviaSET oxidation by the excited photocatalyst MB+*.The 3-aminoakyl chromones56could be obtained by similar photoredox decarboxylation ofN-arylglycine22A,oxidation of aminoalkyl radicals22D,Mannich reaction,intramolecular nucleophilic cyclization and subsequent amine elimination.
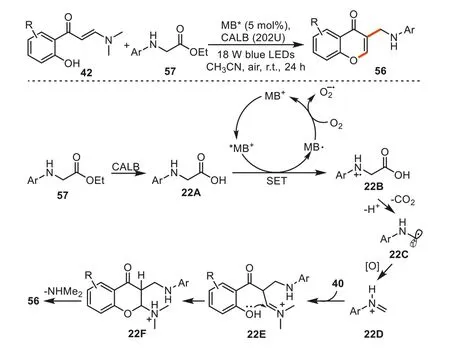
Scheme 22.The combined enzyme and photoredox catalysis for the synthesis of 3-aminoalkyl chromones.
In 2022,Yang and Xiangetal.established a new cascade protocol for the assembly of 3-aminochromones42witho-hydroxyaryl enaminones andO-perhalopyridin-4-yl hydroxyl amine58,a versatile amidyl-radical precursor [94].This protocol featured with mild reaction conditions,synthetic simplicity,and scalability.In the reactions,the photocatalystfac-Ir(ppy)3was first excited to generate its active speciesfac-Ir(ppy)3*under the blue LEDs irradiation.The subsequent quenching of the exited photocatalyst by enaminone42generated the nitrogen-centered radical23Aand Ir2+viaSET process.The highly reactive Ir2+was immediately oxidized by58to furnish amido radical ·NHBoc along with regeneration of the Ir-photocatalyst.A facile radical transposition of23Adelivered the isomeric carbon-center radical23B.The species could be quickly trapped by amido radical ·NHBoc to give intermediate23Cprior to the cyclization process (path a).Alternatively,the cyclic radical23Dmight be formedviaintramolecular cyclization of23Bunder in the presence of base (path b).Radical cross-coupling of23Dwith ·NHBoc led to the intermediate23E.The final products59were providedviathe elimination of dimethylamine (Scheme 23).

Scheme 23.The synthesis of 3-aminated chromones from ohydroxyarylenaminones.
4.3. Synthesis of seven-membered O-heterocycles and carbocycles
In 2018,Yang and Chen developed a practical protocol accessing benzoxepines61/62by using functionalized enaminones60and ethyl bromodifluoroacetate43as starting materials with white LEDs irradiation in the presence of Ir(dtbbpy)(bpy)2PF6[95].In the formation of these fused seven-membered scaffolds,the excited state Ir(III)*occurred from Ir(III) under visible-light irradiation.The further reaction with BrCF2COOEt43gave Ir(IV)+species and RFradicals24A.Subsequently,the free radical addition of24Ato the olefin unit in substrate60and a sequential radical addition cascade process generated radical intermediate24C.The quick oxidation by Ir(IV)+yielded iminium intermediate24Dand regenerated Ir(III).WhenN,N-dimethyl enaminones (R2/R3=Me) were used,the intermediate24Dwas quickly hydrolyzed by water to furnish products61(path a).In the case of the monosubstituted iminium ion (R3=H),the deprotonation and tautomerization processes took place to afford the corresponding enamine62(path b)(Scheme 24).
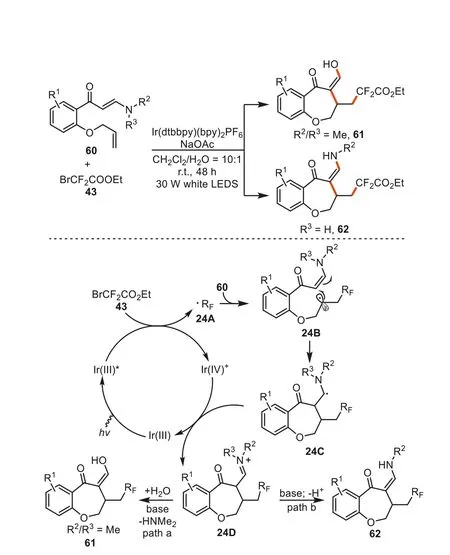
Scheme 24.The photocatalytic synthesis of CF2-containing benzoxepine derivatives.
In 2021,Yangetal.developed a method for the synthesis of cyclohepta[b]indoles64viahighly functionalized enaminones63[96].This approach involves a visible-light-induced cascade of[2+2] cycloaddition and retro-Mannich-type reaction of enaminones.As shown in Scheme 25,radical cation intermediate25Awas first formed by single electron oxidation of substrate63in the presence of excited-state FCNIrpic ([IrIII]*).The oxidized enaminone moiety in intermediate25Areadily underwent radical addition to the indole ring to furnish benzylic radical intermediate25Bwhich then added to the iminium moiety to deliver radical cation25C.The resulting intermediate25Cwas unstable and tended to either go back to25B(breaking bond a) or forward to25D(breaking bond b).The species25Dwould subsequently be reduced by previously generated [IrII] species to zwitterion25E.The final products64were affordedviaan intermolecular proton transfer in25E.
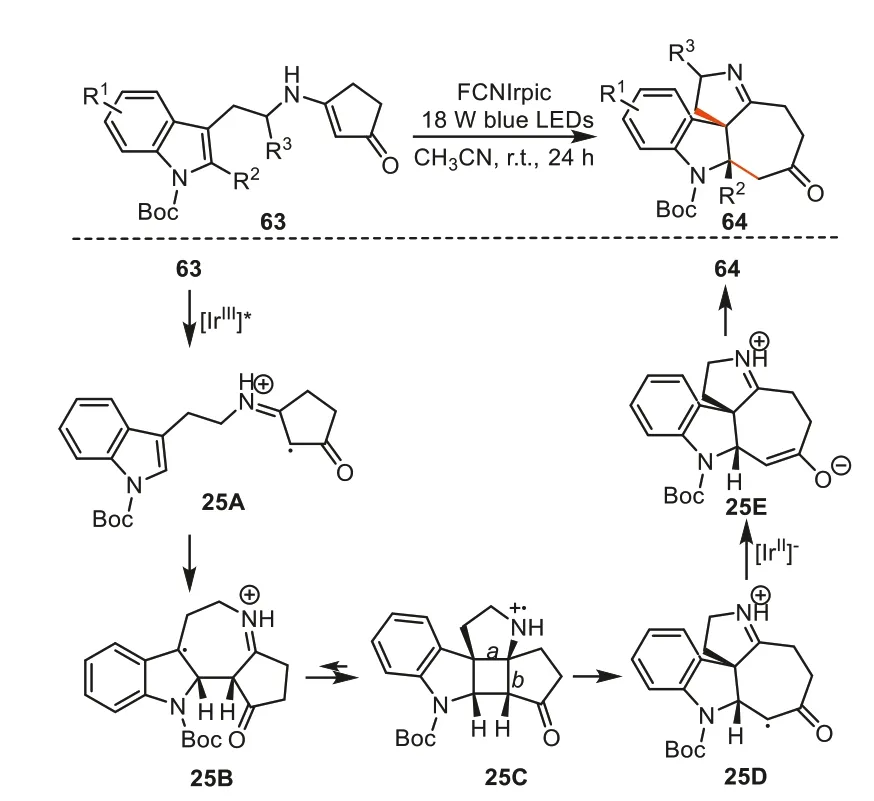
Scheme 25.The photocatalytic synthesis of cyclohepta[b]indoles.
5.Multicomponent reaction for heterocycle construction
Multicomponent reactions (MCRs) are powerful tools in current organic synthesis due to their irreplaceable advantages in simple starting materials,step economy,product diversity and fast construction of molecular diversity [97,98].Designing enaminonebased MCRs with photocatalyst would inarguably benefit the synthetic chemistry by providing highly sustainable routes toward highly diversified organic products.
5.1. Synthesis of pyrimido[1,2-b]indazole derivatives
In 2021,Wang and co-workers reported a visible-light-driven three-component cyclization for the synthesis of pyrimido[1,2-b]indazole derivatives66from enaminones27,3-aminoindazoles65and ethyl bromodifluoroacetate43[99].The reactions were proposed to start from a metal-to-ligand charge transfer (MLCT)process from the photocatalystfac-[Ir3+(ppy)3] to generate excited state IrIII*under blue LEDs irradiation,enabling the C-Br bond cleavage of in43viaSET reduction process to give alkyl radicals(·CF2COOEt).The alkyl radicals subsequently added to the electronrich olefin in the enaminone moiety and led to carbon radicals intermediate26A.The further oxidation on this intermediate by IrIVaccessed to imine cation26BSubsequently,the intermediate26Bunderwent an elimination of HF to afford 1,3-vinylimine ion intermediate26Cwhich was trapped by 3-aminoindazolesviaadditionelimination process (SNV reaction) to form intermediate26Dor its isomer26E.Afterwards,the intramolecular addition of the nucleophilic NH group to the iminium site gave tricyclic intermediate26F.The elimination of dimethyl amine from the amino dihydropyrimidine ring provided products66(Scheme 26).

Scheme 26.Visible-light-driven three-component cyclization for the synthesis of pyrimido[1,2-b]indazole derivatives.
5.2. Synthesis of 2,3-difunctionalized quinolines
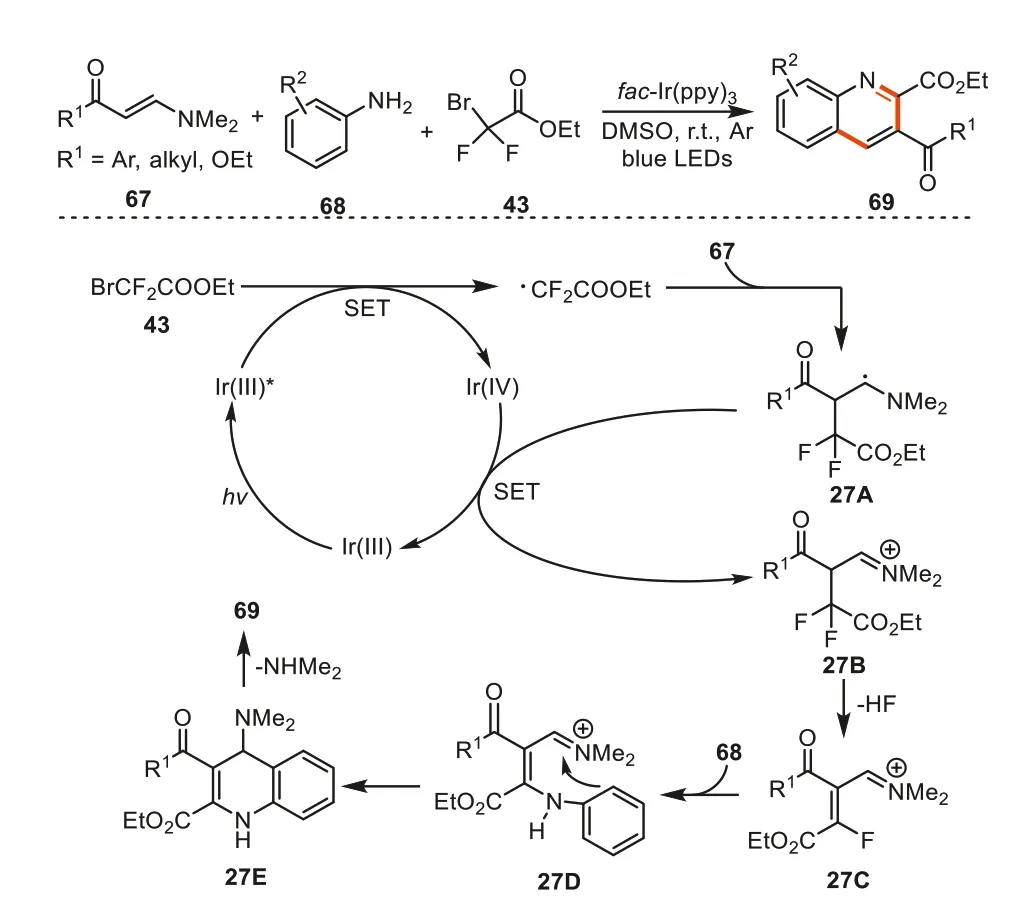
In 2022,Wang’s group reported another three-component protocol for the synthesis of 2,3-difunctionalized quinolines69viathe reactions of enaminones67,aryl amines68and ethyl bromodifluoroacetate43with the irradiation of blue LEDs in the presence offac-Ir(ppy)3[100].The reaction mechanism was familiar with the former one in Scheme 26.The difference was that when 1,3-vinyl imine ion intermediate27Cwas formed,it was rapidly trapped by aryl amine to give intermediate27Dwhich underwent an intramolecular cyclization and amine elimination to provide products69(Scheme 27).
Scheme 27.Photoinduced three-component cyclization for the synthesis of 2,3-difunctionalized quinolines.
5.3. Synthesis of trisubstituted pyrazoles
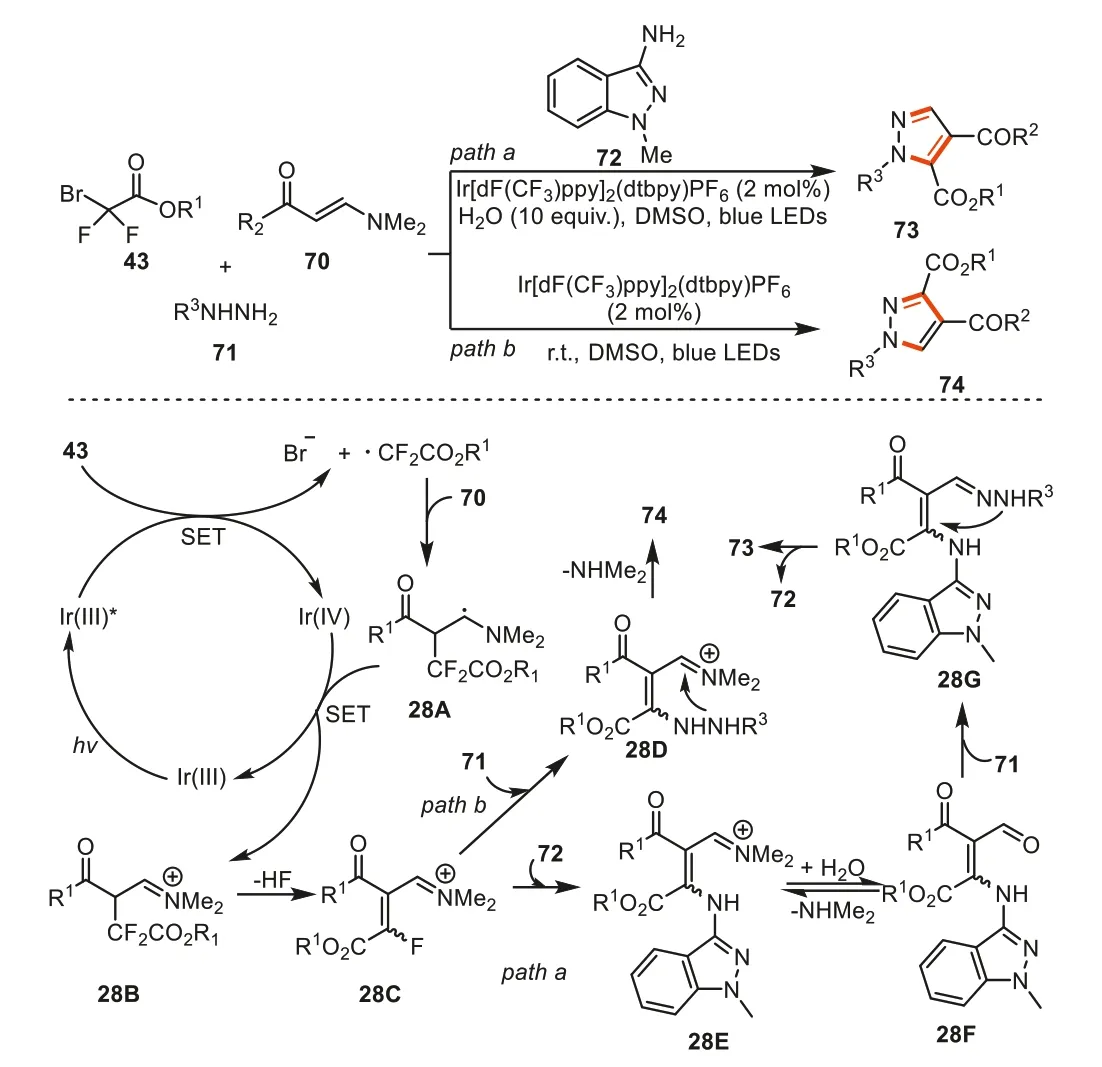
Recently,Wang and Gengetal.again developed a multicomponent method for the tunable synthesis of pyrazoles73and74by employing bromodifluoroalkyl acetates43,enaminones,and hydrazines as starting materials with blue LEDs photocatalysis [101].In these reactions,1-methylindazol-3-amine acted as a traceless mediator in switching the regionselectivity of 1,3,4-trisubstituted and 1,4,5-trisubstituted pyrazole formation.Mechanically,the active 1,3-vinylimine ions intermediate28Cwas first formed through similar transformation processes as in the above works.This intermediate was then trapped by 1-methylindazol-3-amine72to form28E.Intermediate28Emight further undergo hydrolysis to deliver intermediate28F,and subsequently reacted with hydrazine to provide28G.The sequential intramolecular cyclization/deamination processes then took place to afforded 1,4,5-trisubstituted pyrazoles73.On the other hand,when the traceless mediator72was not added,R3NHNH2was directly captured 1,3-vinylimine ions28C,giving intermediate28Dviaan SNV process.The similar intramolecular cyclization/deamination process then led to the formation of 1,3,4-trisubstituted pyrazoles74(Scheme 28).
Scheme 28.Photoinduced switchable multicomponent cyclization for the synthesis of adjustable multi-substituted pyrazoles.
5.4. Synthesis of spiro[indoline-3,4′-quinoline] derivatives
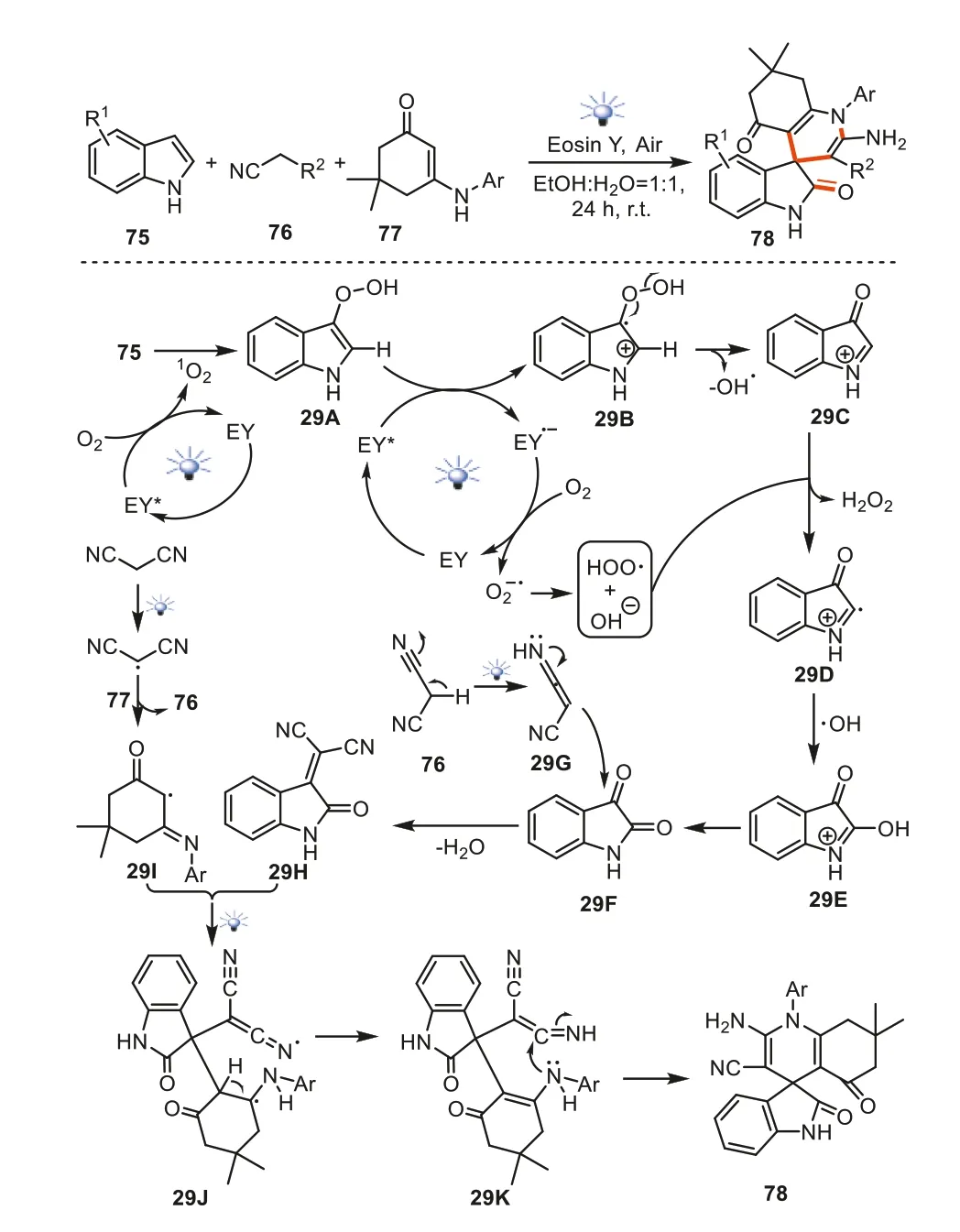
Very recently,Singhetal.reported the visible light-mediated three-component synthesis of spiro[indoline-3,4′-quinoline]derivatives78viathe reactions of indoles75,methylene nitriles76and enaminones77under the irradiation of a 22 W LED lamp [102].In the formation of these spiroheterocyclic products,the initially irradiation of light to Eosin Y led to the excited state EY*which promoted the transformation of molecular oxygen to singlet oxygen.Indoles captured the singlet oxygen to afford peroxo species29A,and a subsequent oxidation by EY*led to29BBy the O-O homolytic cleavage in29B,the cation29Cand hydroxyl radical were created.The C2-site hydrogen atom in29Ccould be abstracted by the peroxyl free radical to deliver intermediate29D.The rapid coupling of29Dwith hydroxyl radical gave29E,and29Ecould further isomerize into29F.Meanwhile,the light irradiation to substrate76led to intermediate29G.The reaction of29Fwith29Gprovided29H.On the other hand,the free radical intermediate29Iwas formedviathe promotion of malononitrile and visible light irradiation.The addition of29Ito29Hled to the formation of29J.After a sequential hydrogen radical transfer and intramolecular cyclization process,the final products78were provided (Scheme 29).
Scheme 29.Visible light triggered three-component cyclization for the synthesis of spiro[indoline-3,4′-quinoline].
6.Conclusions and perspectives
Herein,we reviewed for the first time the advances in the visible light-mediated chemical transformations of enaminones.The modes of transformations,including the direct C(sp2)-Hα-functionalization,C=C double bond functionalization,annulation toward heterocyclic and carbocyclic scaffolds,as well as enaminone-based multicomponent reactions have been covered.These interesting results illustrate the power of combining enaminone chemistry and photocatalysis in the designation and application of energy economic,mild and diversity-oriented organic reactions.However,challenges remain yet in this research area.For example,the frequent application of noble-metal photocatalyst,limited generality in the application of different free radical reaction designation with most of the currently known model,and switchable control of the site selectivity in analogous sites such as the two vinyl C-H bonds are yet non-negligible restrictions.Therefore,in developing more novel and applicable photocatalytic enaminone-based synthesis,extensive efforts are still highly desirable.The discovery on more novel and different transformation and catalytic modes,tuning the chemo-selectivity,application in other important synthesis such as asymmetric synthesis,targetoriented synthesis,are directions worthy of more attention.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors thank the National Natural Science Foundation of China (No.21702088) and the Natural Science Foundation of Shandong Province (No.ZR2022MB130) for the financial support.
杂志排行

Chinese Chemical Letters的其它文章
- The 3rd Xihua Chemistry and Biomedicine Forum
- Professor Hualiang Jiang: A tribute to an esteemed visionary chemist and pharmacist
- Development of porphyrin-based fluorescent sensors and sensor arrays for saccharide recognition
- Recent advances of versatile reagents as controllable building blocks in organic synthesis
- Synthetic host-guest pairs as novel bioorthogonal tools for pre-targeting☆
- Functional nanoemulsions: Controllable low-energy nanoemulsification and advanced biomedical application