深度学习在基于序列的蛋白质互作预测中的应用进展
2024-03-22朱景勇李钧翔李旭辉张瑾毋文静
朱景勇,李钧翔,李旭辉,张瑾,毋文静
(1 浙江理工大学生命科学与医药学院,浙江 杭州 310018; 2 嘉兴学院生物与化学工程学院,浙江 嘉兴 314000;3 浙江清华长三角研究院,衰老科学创新研发中心,浙江 嘉兴 341001; 4 禾美生物科技(浙江)有限公司,浙江嘉兴 341001; 5 浙江清华长三角研究院,浙江省应用酶学重点实验室,浙江 嘉兴 314006)
蛋白质作为细胞中最常见的生物大分子,在细胞内的生物过程中发挥着至关重要的作用。从信号转导、基因的复制、转录与翻译,到细胞能量代谢,都需要蛋白质的参与。很多蛋白质通过与其他蛋白质相互作用来发挥功能[1],因此深入研究蛋白质相互作用(protein protein interactions, PPI)对于了解生命机制、发现药物靶点具有重要意义。目前PPI鉴定方法一般分为传统实验方法和计算方法两种,常见的实验方法有酵母双杂交[2]、串联亲和纯化[3]、蛋白质芯片[4]等。尽管这些方法已经取得了重要成果,但存在成本高、耗时长、假阳性率高等问题[5],而计算方法作为传统实验方法的重要补充,可以提高PPI鉴定的准确性和效率。
PPI预测的传统计算方法一般包括基于基因组信息的方法[6]、基于进化信息的方法[7]、基于蛋白质互作网络的方法[8]以及基于物理模型的分子对接方法[9]等。基于基因组信息的方法通过蛋白质编码基因的同源性、共表达模式和共定位信息推断可能的互作关系,需要大量关于基因的先验知识;基于进化信息的方法通过分析蛋白质序列在多物种中的共进化信号来预测它们之间的相互作用,高度依赖可用进化数据的数量和质量,且这种方法建立在保守进化的基础上,这在某些情况下并不成立;基于蛋白质互作网络的方法根据网络拓扑结构和模块化特性,从已知互作网络关系中推测新的相互作用。近年来常利用图神经网络[10]做这方面的研究,但这种方法完全依赖于已知的互作网络,可能会由于现有蛋白质互作网络并不全面而出现遗漏和假阳性;最后,基于分子对接的方法在分子级别上模拟蛋白质与潜在合作伙伴之间的结合,这类方法具有明确的生物物理基础,通过结合自由能来判断结合状况。但分子对接通常需要蛋白质三维结构信息,且计算复合物的构象伴随着复杂的计算过程,需要巨额的算力和时间。
随着大数据和计算能力的发展,利用蛋白质信息进行训练和预测的机器学习(machine learning)方法已在PPI预测领域取得快速进步。高通量技术的发展产生了大量PPI数据[11],这为机器学习在PPI预测上的应用奠定了坚实的数据基础。目前常见的基于机器学习的PPI预测方法可以分为基于结构信息的预测和基于序列信息的预测。基于结构信息的计算方法通常依赖蛋白质结构数据进行PPI预测,如Inpred[12]和Struct2Graph[13]等,但这类方法仅适用于有确定三维结构信息的蛋白质。然而传统测定蛋白质三维结构依赖X射线晶体学和核磁共振等方法,这类方法通常耗时且昂贵[14]。相较于蛋白质的三维结构,高通量技术带来了大量蛋白质的序列信息,尽管科研人员一直致力于测定序列的三维结构,但已知结构的序列和未测定结构的序列在数量上存在显著差距[15],这种数量差异也是基于序列的方法成为研究热点的重要原因。最早在2007年,Shen等[16]首次提出了仅基于序列的PPI预测模型,使用支持向量机作为分类器,最终达到了83.9%的准确率,展示了机器学习算法在基于序列信息预测PPI领域的潜力。随着技术的发展,基于决策树[17]、朴素贝叶斯[18]、随机森林[19]等算法的预测模型都取得了进展。
然而,传统机器学习方法预测PPI需要研究者根据专业知识给每条蛋白质序列手动生成特征[20],可能会遗漏蛋白质信息中难以捕捉的深层特征,这限制了其在预测蛋白质互作时的准确性和泛化能力。而深度学习(deep learning)方法已经在这方面展现了巨大的潜力,作为机器学习的分支,深度学习已经在计算机视觉[21]和自然语言处理[22]等领域成功应用。通过利用多层非线性处理单元进行特征提取和转换,深度学习模型可以从庞大而复杂的数据集中学习分层表征,这一特点在计算生物学中非常有用[23]。近年来学者将其应用于PPI预测,依靠强大的学习能力,深度学习实现了从蛋白质信息中抽取深层特征,提高PPI预测的准确性和效率。利用深度学习进行PPI预测一般流程包括数据准备、序列编码、算法学习和预测输出(图1)。
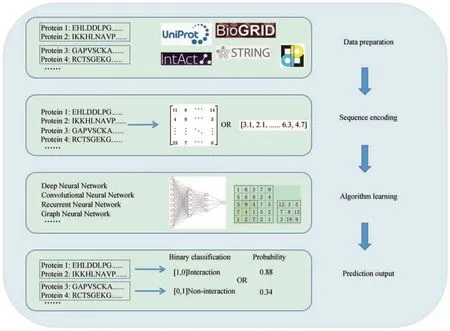
图1 利用深度学习基于序列预测蛋白质相互作用的一般流程Fig. 1 General workflow for predicting protein-protein interactions by sequence-based deep learning
随着时间推移,深度学习在基于序列预测PPI的应用上快速发展,总体可分为三个方向:预测蛋白质之间是否互作;预测蛋白质序列上的结合位点;预测蛋白质间形成的复合物结构。本文旨在综述深度学习在基于序列的蛋白质互作预测领域的应用,主要聚焦于预测蛋白质之间互作可能性的模型,提到了部分预测蛋白质序列上结合位点的模型。本文涵盖计算模型中数据集的构建方法、常用蛋白质编码方法、常用的算法框架以及模型评估指标等方面。同时,我们还将分析目前所面临的挑战和未来发展趋势,以期为该领域的研究提供参考。
1 PPI预测模型中的数据处理
1.1 数据集的构建
深度学习方法是数据驱动的,高质量的数据对于深度学习模型至关重要。数据集越大越全面,训练好的模型就越能适应可能出现的新数据,有利于提高预测的准确性。对于基于序列的PPI预测,数据集主要由相互作用的蛋白质对(阳性数据)和非相互作用的蛋白质对(阴性数据)组成。研究人员们通常从已公开发表的数据库提取数据,本节将介绍常用的相关数据库,以便研究人员在进行相关研究时能够更好地选择和使用。
蛋白质序列一般从UniProt[24]中提取,这是一个包含蛋白质序列和相关注释信息的综合性数据库,其中大部分信息通过高通量测序技术获得。这些蛋白质来自人类、动物、植物、微生物等不同物种,涵盖了广泛的研究领域。除了序列信息外,UniProt还包含了各种类型的蛋白质注释信息,包括功能、结构、组织特异性、代谢途径、互作信息等。
相互作用对的数据一般通过公开发表的互作数据库获取,BioGrid[25]是一个蛋白质相互作用数据库,目前包含了来自7万多份已发表文献的17万多份互作信息,涵盖包括酵母、蠕虫、苍蝇、老鼠和人类在内的多种物种。HPRD[26]是一个以人类蛋白质为核心的蛋白质相互作用信息数据库,涵盖了超过30 000个人类蛋白质。DIP[27]包含了各种物种的蛋白质互作信息,其中人类蛋白质互作信息达9000多对。
除了相互作用数据外,非相互作用数据对于模型训练同样重要,常见的生成方式为打乱互作蛋白质对,并将其随意组合。已经有研究证明[28],这种方式得到的蛋白质对的互作可能性可以忽略。另外,部分研究者们通过Negatome[29]数据库获取非互作对数据,Negatome通过筛选文献和分析已知三维结构的蛋白质复合物,并从中排除高通量方法检测到的相互作用来收集非相互作用蛋白质对。一些常见的互作数据库及基本信息如表1所示。

表1 常用的数据库以及基本信息Table 1 Public databases and basic information
1.2 蛋白质序列的编码
尽管深度学习具有自动提取特征的能力[34],但在基于深度学习的PPI预测方法中,将蛋白质序列编码为矩阵或者向量仍有必要。一般而言,天然蛋白质是由20种标准氨基酸组成的长度不等的序列,而计算模型只能接受数字向量作为输入[35],所以在深度学习方法中,需要将这些离散的氨基酸符号转换为连续的数值表示,并进一步表示为固定的矩阵或者向量,使其更容易被深度学习模型处理。高效的编码方式可以帮助模型关注PPI中更有意义的信息,或使计算模型更全面地掌握蛋白质信息以及互作信息,从而更好地学习到互作“规律”,提高模型的预测性能。本节将介绍现有基于序列的计算模型中常用的蛋白质序列编码方法,主要分为基于序列成分、基于自相关和基于进化信息的编码方法。一些常见的蛋白质编码方法如表2所示。

表2 基于序列的PPI预测模型中常见的蛋白质编码方法Table 2 Protein encoding methods in sequence-based PPI prediction models
1.2.1 基于序列成分的编码方法
基于序列成分的编码方法通过计算序列中氨基酸或连续氨基酸对的出现频率来表示一条蛋白质序列。最早,研究人员通过独热编码(one-hot)[50]来表示一条序列,将20个标准氨基酸按一定顺序固定,对于序列中第i位氨基酸用20个二进制位表示,第i位设为“1”,其余为“0”,对于一条长度为N的序列,最后能得到N×20的矩阵(见图2)。这种方法虽然能将氨基酸序列转换为矩阵,但数据过于稀疏,且长序列的独热编码存在维度过高的缺点,容易导致计算效率低下。在独热编码思想的基础上通过计算蛋白质序列内20个标准氨基酸的出现频率得到氨基酸组成(amino acid composition, AAC),最终对于每一条蛋白质序列都可以得到一个20维的向量。联合三元组将连续三个氨基酸看成一个三元组,通过计算三元组的频率表征序列信息,这种方法可以关注相邻氨基酸之间的相互影响。伪氨基酸组成(pseudo-amino acid composition, Pse-AAC)在AAC的基础上结合了氨基酸的疏水性、亲水性和侧链质量,并考虑了序列顺序信息,使编码后的向量包含更丰富的信息。

图2 利用one-hot将氨基酸序列转化为向量矩阵Fig.2 Converting amino acid sequences into vector matrices using the one-hot strategy
除序列的基本信息外,蛋白质的内在无序区域也与PPI密切相关。蛋白质的内在无序性描述的是蛋白质序列中某些区域在没有与其他分子或蛋白质相互作用的情况下,不倾向于形成固定的三维结构[51]。这些无序的区域在许多生物学过程中起着关键作用,特别是在蛋白质互作中[52]。已有研究表明,蛋白质的无序区域通过短线性基序和无序区域的诱导折叠介导PPI[53]。具体地说,一个蛋白质的短线性基序经常位于其序列的无序区域,并通过与伴侣蛋白质的结构域进行互作来实现PPI[54]。某些无序区域还会通过诱导折叠与另一个蛋白质的结构域或无序区域结合[55]。另外,由于这些无序区域没有固定的结构,它们常常作为蛋白质互作网络中的枢纽,扮演核心的调节角色[56]。因此,已经有研究人员利用工具预测蛋白质中每个残基的无序倾向,并将这些无序倾向作为特征向量引入到PPI预测模型的训练中[57]。通过计算方法预测蛋白质的无序区域是近年来的热门研究方向,常用的预测工具包括IUPred2[58]、DEPICTER[59]以及在CAID(Critical Assessment of protein Intrinsic Disorder prediction)[60]竞赛中取得第一名的SPOTDisorder2[61]等。CAID竞赛是评估和促进蛋白质无序倾向预测方法发展的重要平台,对推动蛋白质无序倾向预测方法的进步具有重要意义。SPOT-Disorder2由Zhou等人提出,通过结合深度挤压和激励剩余开始(Squeeze-and-Excitation residual inception)神经网络[62]与长短时记忆(long short-term memory, LSTM)[63]神经网络,综合利用进化信息和序列属性,显著提高了预测精度。对于输入序列的每一个残基,SPOT-disorder2都会给出一个0到1的值代表该残基倾向无序的概率,这些数值可以被编码为特征向量送入PPI模型中训练。类似的编码如IUPred2,提供一条长度为L的蛋白质序列即可获得一个L×3的矩阵,包含了这条蛋白质的氨基酸组成和IUPred2预测每位残基倾向无序的概率。
1.2.2 基于自相关信息的编码方法
基于序列成分的编码方法重在关注序列中氨基酸的种类和频率,且最多只考虑相邻氨基酸之间的相互影响,但PPI可能发生在不连续的氨基酸片段,所以不相邻氨基酸之间的相关性至关重要。基于这一点,Guo等[39]最早提出了自协方差(auto covariance, AC)用来计算序列中各个位置氨基酸的相关性,首先根据氨基酸的七种理化性质值将蛋白质序列转换为数值序列,然后通过自相关公式计算得到各个位置氨基酸之间的自相关性作为特征向量进行下一步的预测。物理化学距离变换(physicochemical distance transformation, PDT)[44]选择了531种理化性质值将氨基酸转换为数值表示,再计算不同距离的氨基酸之间的相关性来表示氨基酸序列。这类方法还包括交叉协方差(cross covariance, CC)[40]以及自交叉协方差(auto-cross covariance, ACC)[41]等,其中氨基酸的理化性质可以通过AAindex[64]获取。这类方法通过计算序列中氨基酸的自相关性来捕捉序列中不同位置氨基酸之间的相互影响,以帮助揭示序列中潜在的相互作用,比基于序列成分的编码方法更全面。
1.2.3 基于进化信息的编码方法
基于进化信息的编码方法旨在通过蛋白质的进化信息进一步掌握序列中蕴藏的潜在特征。位置特异性得分矩阵(position specific scoring matrices,PSSM)[45]是其中最常见的方法,将蛋白质序列通过PSI-BLAST[65]在蛋白质数据库中检索比对,最终生成一个N×20的矩阵,其中N为蛋白质序列的长度,以每个位置的氨基酸概率分布的形式揭示蛋白质序列的进化信息。除PSSM外,基于进化信息的编码方法还包括BLOSUM62[66]、ACC-PSSM[46]、Pseudo-PSSM[67]等,这些方法在以预测蛋白质互作为核心的研究中起到了重要作用。
2 深度学习在基于序列预测蛋白质互作中的应用
2017年,Sun等[68]首次将深度学习技术成功地应用于蛋白质互作预测研究中,他们采用了堆叠自动编码器(stacked autoencoder, SAE)对基于序列信息的蛋白质互作进行预测,在多个外部测试集上的预测准确率达到了87.99%~99.21%,充分展示了深度学习方法在该领域的巨大潜力。如今,深度学习方法在基于序列信息预测蛋白质互作研究中发挥着关键作用。从算法层面看,这些方法可划分为基于深度神经网络(deep neural network, DNN)[69]的方法、基于卷积神经网络(convolutional neural network, CNN)[70]的方法、基于循环神经网络(recurrent neural network,RNN)[71]的方法、基于注意力机制和Transformer[72]的方法以及基于混合神经网络的方法。本节将分别介绍这些算法架构及其在基于序列的蛋白质互作预测中的代表性应用,表3总结了近5年的相关计算模型,并按照算法框架及发表时间做了分类和排序。
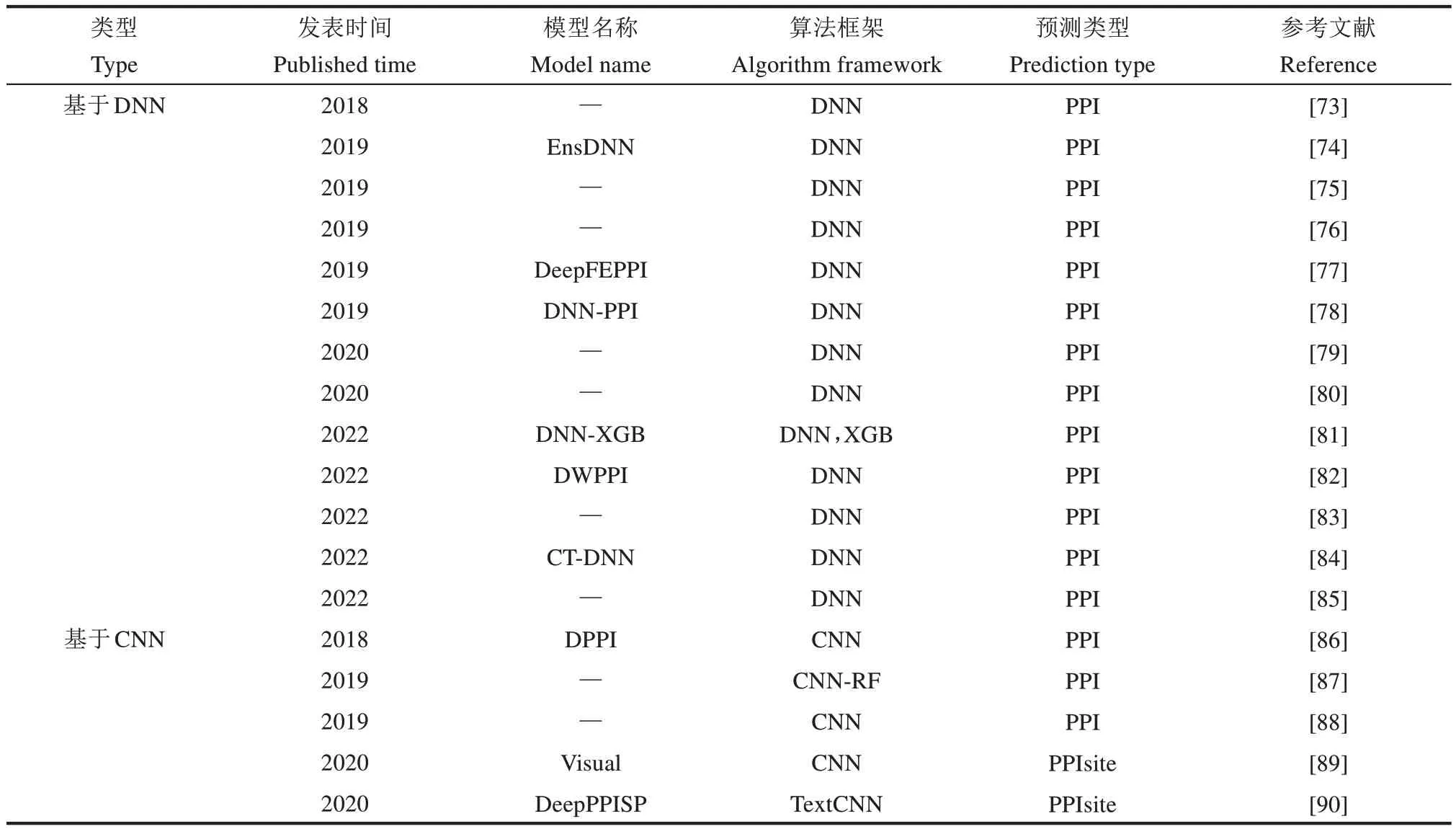
表3 近5年相关的计算模型Table 3 Computational models developed within the past 5 years
2.1 基于深度神经网络的方法
深度神经网络是一种基于多层感知器(multilayer perceptron, MLP)的前馈神经网络,包括输入层、多个隐藏层和输出层[图3(a)]。DNN通过多个隐藏层对输入层的数据进行层层计算,最后通过输出层进行决策,如通过softmax等激活函数进行二元分类。在蛋白质互作研究中,DNN可以通过多层网络提取氨基酸序列信息中的深层特征,学习蛋白质互作的复杂模式,从而更精准地预测蛋白质之间的相互作用。

图3 DNN、RNN、CNN的基本网络结构Fig. 3 Basic network structure of DNN, RNN, and CNN
2017年Du等[118]基于DNN构建了一个名为DeepPPI的计算模型,他们从蛋白质序列中提取包括氨基酸组成在内的五种描述符,通过两个独立的深度神经网络分别处理一对样本中的每条序列,最后对PPI进行预测,在测试集上取得了92.5%的准确率。2019年Yao等[77]提出DeepFEPPI模型,通过Res2vec方法表示蛋白质序列,同样使用两个独立的深度神经网络分别提取每个蛋白质的深层特征,最后通过softmax函数对PPI进行分类,在测试集上取得了98.71%的准确率。2021年Mahapatra等[81]提出了名为DNN-XGB的计算模型,通过氨基酸组成、联合三元组、局部描述符[119]表示序列,使用深度神经网络提取特征最后利用极端梯度提升进行PPI的分类,在测试集中最高达到了99.74%的准确率。
基于深度神经网络的计算模型在蛋白质相互作用预测方面取得了显著的成果。DNN模型强大的特征提取能力使其能够捕捉蛋白质序列中的复杂特征。然而,DNN在捕捉蛋白质序列中的局部特征仍有局限性,且多层数提高特征提取能力的同时存在着计算量过大的问题,部分研究人员开始探索基于卷积神经网络的计算模型。
2.2 基于卷积神经网络的方法
卷积神经网络作为深度学习算法的一个分支,通过卷积层、池化层和全连接层的组合以实现对输入特征的自动提取和分类,可以自动提取局部特征,然后经过池化层实现特征降维,使网络具有更强的表达能力和泛化能力,其基本结构见图3(c)。相较于DNN,CNN减少了全连接层的数量,从而减少了参数数量,大大降低了计算复杂度。在PPI预测的应用中,CNN可以高效地捕捉蛋白质序列中的局部依赖关系,从中学习到与互作相关的特征,做到更精准预测。
2018年Hashemifar等[86]提出了一个名为DPPI的框架,通过包括卷积、随机投影、预测在内的三个模块仅根据序列信息对PPI进行预测,在酿酒酵母数据集上最高达到了96.68%的准确率,且在同一测试集下的查全曲线下面积优于其他算法。2020年Li等[91]提出了EnAmDNN方法,通过局部描述符、自协方差、联合三元组、伪氨基酸组合来编码蛋白质序列,利用多层卷积神经网络自动提取蛋白质特征,并结合自注意力机制来分析蛋白质之间的深层关系,最终在5个独立数据集上最高达到了94.67%的准确率。2021年Lei等[57]提出了CAMP方法用于蛋白质-多肽的相互作用预测,通过长度限制来区分蛋白质和多肽,模型规定蛋白质的长度≤800个氨基酸,而肽的长度≤20个氨基酸。该方法利用卷积神经网络结合自注意力机制分别提取给定多肽和蛋白质信息中的特征进行互作预测,同时识别多肽序列上的结合位点。2022年,Hu等[97]提出了DeepTrio方法,使用一个基于掩模多个并行CNN提取蛋白质序列的多尺度上下文信息进行PPI预测,在多个测试集上取得优秀效果。2023年Gao等[98]开发了EResCNN方法,首先通过PseAAC、AC、Pse-PSSM、EBGW、MMI、CT方法提取每对样本中每条蛋白质序列特征,然后利用三层卷积层和三层池化层逐层学习潜在特征,最后结合XGBoost、RF、LightGBM和极度随机树对PPI进行预测分类,在测试集中达到了最高98.61%的准确率,展示了深度学习和传统机器学习结合的潜力。
基于卷积神经网络的计算模型在蛋白质相互作用预测中取得了显著的成绩,但在捕捉长距离依赖关系方面仍然面临挑战,而循环神经网络模型及其变体在这方面具有一定优势,并取得了一些成果。
2.3 基于循环神经网络的方法
循环神经网络是一类具有内部状态短期记忆能力的网络结构,通过循环连接使网络可以在处理序列后部分数据时保留之前的信息,能够捕捉输入序列中的长程依赖关系,在处理序列数据方面具有显著优势,其基本结构见图3(b)。蛋白质序列中的氨基酸之间通常具有相互依赖关系,所以RNN在基于序列的蛋白质互作预测中已有部分研究。然而传统的RNN容易出现梯度消失或梯度爆炸问题,这限制了它们在处理长序列时的性能,为了克服这些问题,研究人员提出了一些RNN的变体,如长短时记忆网络[63]和门控循环单元(gated recurrent unit, GRU)[120]。这些变体通过引入门控机制来调控信息在网络中的传递,从而改善了网络在处理长序列时的性能。RNN及其变体可以捕捉蛋白质序列中的局部和全局依赖关系,从而有助于揭示潜在的互作模式。因此,在接下来的部分中,我们将重点介绍基于RNN及其变体的模型在基于序列的蛋白质互作预测中的应用。
2018年Gonzalez-Lopez等[100]提出了一种基于RNN和嵌入技术的方法,不依赖手动特征工程,直接对原始氨基酸序列进行处理,最终在测试集中最高达到了92.59%的准确率,体现了RNN在该方面研究的潜力。2019年,Zhang等[101]提出了一个名为DLPred的方法,该方法基于一个简化的长短时记忆网络,利用PSSM、理化性质、亲水性指数等特征对蛋白质互作位点进行预测。Alakus等[102]提出了一个基于LSTM的方法,首先通过蛋白质标记和ProtVec方法将蛋白质序列转化为数字,随后通过LSTM对PPI进行预测,最高达到了92%的准确率。2023年Mewara等[106]提出了一个基于双向长短时记忆网络(bidirectional LSTM,BLSTM)的方法,直接从原始氨基酸序列中提取特征并进行PPI预测,最终在幽门螺杆菌数据集上达到了99.54%的准确率。
尽管RNN在捕捉长距离依赖关系方面做出了重要贡献,且LSTM和GRU等RNN变体有效地解决了传统RNN的梯度消失问题,但在处理长序列和捕捉复杂的互动模式方面,以循环神经网络为主体的框架仍然存在计算效率的挑战。
2.4 基于注意力机制和Transformer的方法
为了更高效地处理长序列并深入解析蛋白质间的复杂相互作用模式,研究者们转向了注意力机制和Transformer。注意力机制的基本思想见图4(a),当模型进行预测时,它可以“聚焦”于输入信息中的某些部分,而忽略其他不相关或不重要的部分。这种“聚焦”是基于输入数据的内容和当前的任务来决定的,从而允许模型在处理长序列时更有选择性地分配其计算资源。这一特性使其在长序列任务中表现出色。Transformer则是一种基于注意力机制的网络架构[图4(b)],能够并行处理输入序列中的所有元素,而不是像RNN那样逐个处理。这种并行处理的能力不仅加速了训练,还使模型能够捕获序列中的长距离依赖关系。Transformer的设计避免了传统的循环或卷积操作,通过引入位置编码来有效地保留和利用序列的位置信息,显著提升了计算效率。

图4 注意力机制和Transformer的基本结构Fig. 4 Basic structure of the basic structure of Attention mechanism and Transformer
近年来,基于注意力机制和Transformer的蛋白质相互作用预测方法取得了显著进展。2022年,Asim等[109]提出了一种名为ADH-PPI的深度混合模型,该模型融合了长短期记忆层、卷积层和自我注意层,其在两个不同物种数据集上的预测准确率均比其他现有方法提高了4%。同年,Li等[110]提出了SDNN-PPI模型,该模型采用氨基酸组成、联合三元组和自协方差对蛋白质序列信息进行编码,并且结合自注意力机制进一步增强了其深度神经网络的特征提取能力,展现出了很好的预测效果。2023年,Nambiar等[121]基于Transformer设计了一个新颖的神经网络架构,称为PRoBERTa。这一架构受到了BERT和RoBERTa训练流程的启发,通过减少Transformer的层数,引入了LAMB优化器来进行模型的优化。在经过微调后,该模型针对PPI预测任务在来自HIPPIE数据库的三个不同数据集上都表现出了超越其他方法的性能,为此类预测任务提供了一个非常有前景的新框架。
2.5 基于混合神经网络的方法
在前面的章节中,我们分别讨论了基于DNN、CNN、RNN及其变体与基于注意力机制和Transformer的深度学习模型在基于序列信息预测蛋白质相互作用方面的应用。尽管这些模型各自具有显著的优势,如DNN的强大表示能力、CNN的局部特征提取能力、RNN的序列依赖性捕捉能力和Transformer的长序列高效处理能力,然而单一类型的网络结构难以充分利用这些优点。为了进一步提高预测性能,研究人员开始探索将不同类型的深度学习模型相互结合,形成所谓的混合模型,以融合多种网络结构的优势,更好地捕捉蛋白质序列的多尺度特征。
2018年Li等[112]提出了DNN-PPI,将两种相互作用的蛋白质序列经过编码和嵌入处理后,依次通过CNN和LSTM神经网络层提取特征,最后将两个输出向量串联起来作为全连接神经网络的输入来预测蛋白质互作,在6个外部数据集上达到了92.80%~97.89%的预测精度。2019年Chen等[113]提出了PIPR方法,PIPR是一种端到端的框架,采用词典嵌入方法对每个蛋白质序列进行编码,然后使用循环卷积神经网络来捕捉编码后的蛋白质序列的特征,最后通过一个多层感知器进行PPI的分类,在基于5倍交叉验证的酵母数据集中达到了97.09%的准确率。2019年Guo等[114]提出了IPPI方法,从AAindex中获取氨基酸属性对蛋白质序列进行编码,通过LSTM和DNN架构对PPI进行预测。综上所述,基于混合神经网络的模型在基于序列信息预测蛋白质相互作用方面取得了显著的进展。通过整合DNN、CNN和RNN等不同类型的深度学习结构,这些混合网络模型成功地充分利用了各自的优势,如强大的表示能力、局部特征提取和序列依赖性捕捉等。这使得混合网络模型在捕获蛋白质序列的多尺度特征和特性方面具有更高的灵活性,从而提高了预测性能。
3 评估指标
深度学习已经在蛋白质互作预测领域取得显著成果,这种技术的成功在很大程度上依赖于我们如何衡量模型的性能。可靠的评估指标可以让研究者客观地了解模型效果从而去调节、优化模型,这就是评估指标的关键性所在。通常,深度学习方法的预测结果可以分为四类,分别是真阳性、真阴性、假阳性和假阴性,它们的具体含义可以参见表4。

表4 四种基本预测结果及其具体含义Table 4 Four basic prediction results and their specific meanings
基于这4种基本预测结果衍生出多种常见的评估指标,其中最常用的有6个,分别是:准确率(accuracy)、精准率(precision)、敏感性(sensitivity)、特异性(specificity)、F1值(F1 score)和马修斯相关系数(Matthews correlation coefficient,MCC),它们的具体计算方法见表5。准确率描述了模型在总体上的预测准确性。精准率反映了预测为阳性的样本中实际为阳性的比例,描述模型预测阳性的准确程度。敏感性,又称为召回率(recall),代表实际为阳性的样本中被正确预测为阳性的比例,体现了模型对阳性的识别能力。特异性代表实际为阴性的样本中被正确预测为阴性的比例,体现模型对阴性的识别能力。F1值为上述精准率和敏感性的调和平均值,为这两个指标提供了一个平衡的度量。马修斯相关系数结合了真阳性、假阳性、真阴性和假阴性四个指标进行评估,值介于-1和1之间,其中:1表示完美预测,0表示预测结果与随机预测一致,-1表示预测结果完全不符合实际。
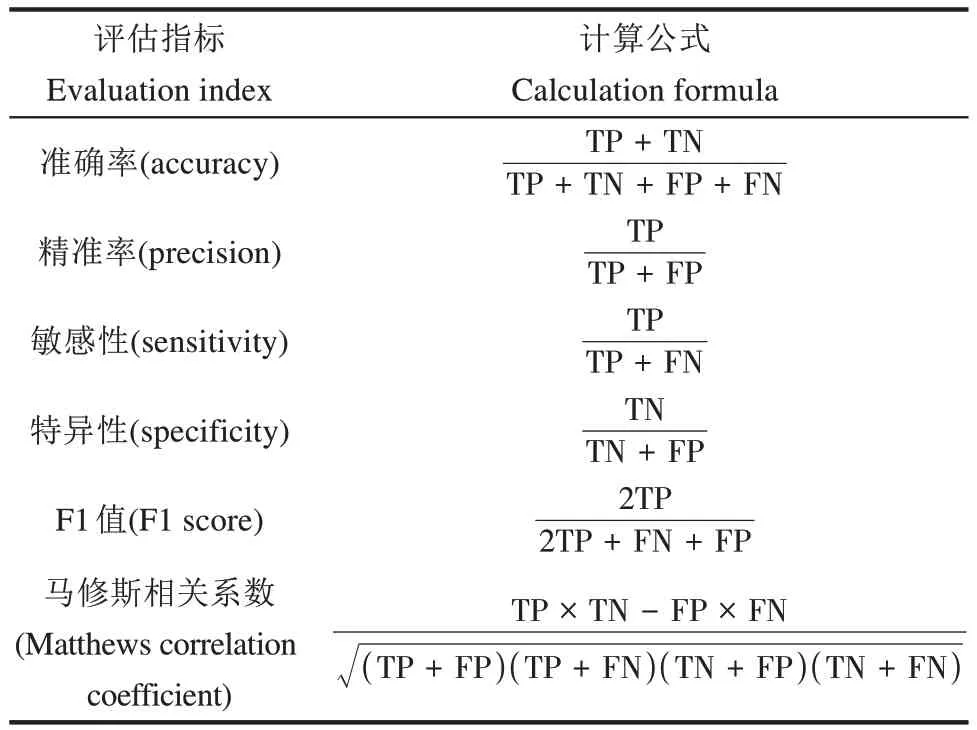
表5 六种评估指标及其计算公式Table 5 Six evaluation metrics and their calculation formulas
在深入研究基于序列的蛋白质互作预测时,许多模型在测试集上的准确率高达90%或95%以上。如此卓越的准确率表面上似乎表明蛋白质互作预测的难题已经接近解决,然而实际上,仅凭准确率或者其他某单一指标,我们可能会忽视其中的一些细微但关键的问题。首先,数据集的来源、预处理方式及实验设计均可能对某一指标产生显著影响。某些模型可能经过针对特定数据集的微调优化,因此在该数据集上表现出色,但在其他数据或实际应用环境中不能维持相同的表现。其次,在实际应用中蛋白质互作的鉴定并不是一个简单的二分类问题,两个蛋白质互作的强度、种类及其在细胞中的具体位置等都是至关重要的。这意味着尽管模型在分类任务上达到了高准确率,但仍可能在实验验证中被证实为假阳性。虽然评估指标有以上的局限性,但是对于模型本身来说仍然是重要且有益的,我们可以根据这些评估指标再尽可能地优化模型,从而更好地辅助PPI鉴定。
综上所述,评估指标是优化模型的关键,理解并正确地看待各项指标能够帮助我们在理论方面明晰优化方向,开发出更高效、准确的预测模型,最终推动我们在蛋白质互作研究中的进步。
4 总结与展望
PPI的鉴定对于许多生物过程的理解至关重要。在本综述中,我们详细讨论了深度学习在基于序列的PPI预测中的应用,介绍了数据集的构建与蛋白质序列的编码方法,重点介绍了基于深度神经网络、卷积神经网络、循环神经网络及其变体和基于混合网络的计算模型,最后介绍了深度学习中常用的评估指标及其计算公式。
深度学习方法已经在PPI预测中取得了显著成果,展示出深度学习技术在该领域的巨大潜力。然而,尽管当前的研究已经取得了一定的进展,但仍然有很多问题有待解决。首先,大多数计算模型都遵循监督学习范式,对训练数据的依赖程度高[122],而当前蛋白质互作数据集存在以下弊端:①主要由实验方法获得,存在假阳性和假阴性,影响了预测的准确性;②数据集覆盖的物种范围较窄,通常来自于酵母或人类,这限制了模型的泛化能力[123]。泛化能力描述的是模型在训练集外的数据上的性能表现,一个具有良好泛化能力的模型能够很好地捕捉到训练数据背后的真实分布,从而在新的数据上也能够做出准确的预测。相反,泛化能力差的模型很可能在遇到新类型数据时表现不佳。因此,模型的泛化能力直接决定了其在实际应用场景中的价值与效果。由于目前用来训练PPI预测模型的数据通常来源于酵母或者人类,这一局限性可能导致模型在处理其他物种时出现预测偏差。因此,建立更大、更广泛、更高质量的蛋白质互作数据集是有必要的。
另外,现有常用的蛋白质序列编码方法仍有局限性,例如本文1.2节提到的方法都是通过简单的线性关系表示蛋白质序列,无法全面地捕捉到蛋白质序列中的内在复杂性。因此,探索更有效的序列表示方法有助于更准确地揭示蛋白质序列的丰富信息和潜在特征。参考当前语言类模型取得了突破性进展[124],基于自然语言处理的方法显示出了巨大的潜力,尽管这些技术最初是为文本和语言设计的,但它们在处理蛋白质序列时也表现出了惊人的能力。词嵌入技术(如Word2Vec和FastText)[125]以及基于Transformer的模型(如ProtBERT)[126]已经成功地利用自然语言处理思想来编码蛋白质序列。这些方法为蛋白质序列提供了一种动态的、上下文相关的表示,与传统的编码方法相比,它们能够捕捉更多的序列模式和特性。
除了数据集的物种来源和蛋白质的序列编码,数据多样性也是值得探索的方向。虽然仅基于序列的模型已经被证明是可行的[68],但考虑到生物系统的复杂性和多样性,序列数据、结构数据、基因表达数据以及其他分子生物学数据都蕴藏着丰富的PPI相关信息。未来可以尝试将多种信息融合进行特征提取进而预测PPI,该策略旨在综合各类数据的优点,为蛋白质之间的复杂互作关系提供一个更全面的视角。通过融合不同数据类型的特性和信息,我们可以构建更为准确和稳健的PPI预测模型。
在本综述的引言部分,我们提到了基于结构的模型由于蛋白质结构的获取成本较高,已测定的蛋白质结构数量不充足,受到发展限制。然而随着AlphaFold2[127]的出现,蛋白质结构预测的准确性显著提高,帮助研究者在没有实验结构数据的情况下获得高质量的蛋白质预测结构。这一技术有助于辅助PPI预测,为基于结构的PPI预测方法开辟了新的道路,值得深入研究。展望未来,随着生物大数据的增长和新型生物信息学技术的出现,多种数据类型的融合预计将成为PPI预测和其他生物信息学任务的核心策略,为该领域带来深刻的变革。
除了上述数据和输入层面的问题,随着深度学习的不断发展,通过设计算法或选择合适的新算法来增强现有深度学习模型的PPI预测能力值得探索。另外也可以考虑结合其他计算方法进行更详实的预测,如将深度学习模型和传统机器学习相结合已经被证明具有潜力。
随着技术的不断发展和计算能力的显著提升,我们有充分的理由相信深度学习将在基于序列预测蛋白质相互作用领域扮演更加核心的角色,有望显著提升PPI预测的准确性和泛化能力,并且能为基于PPI的靶点研究、药物研发和疾病机制探索等相关研究提供有力帮助。同时我们也应注重深度学习技术与生物实验研究的紧密结合,以确保算法开发的科学性和实用性,努力向可解释深度学习方向发展。总之,深度学习在基于序列预测PPI领域的应用,将提升蛋白质相互作用网络的解析效率,推动人类对生命过程本质的认识。
