Hydralazine represses Fpn ubiquitination to rescue injured neurons via competitive binding to UBA52
2024-03-21ShengyouLiXueGaoYiZhengYujieYangJianboGaoDanGengLingliGuoTengMaYimingHaoBinWeiLiangliangHuangYitaoWeiBingXiaZhuojingLuoJinghuiHuang
Shengyou Li , Xue Gao , Yi Zheng , Yujie Yang, Jianbo Gao, Dan Geng, Lingli Guo,Teng Ma, Yiming Hao, Bin Wei, Liangliang Huang, Yitao Wei, Bing Xia, Zhuojing Luo,Jinghui Huang
Department of Orthopedics, Xijing Hospital, Fourth Military Medical University, Xi'an, 710032, China
Keywords:Ferroptosis UBA52 Ferroportin Ubiquitination Hydralazine Peripheral nerve injury
A B S T R A C T A major impedance to neuronal regeneration after peripheral nerve injury (PNI) is the activation of various programmed cell death mechanisms in the dorsal root ganglion.Ferroptosis is a form of programmed cell death distinguished by imbalance in iron and thiol metabolism, leading to lethal lipid peroxidation.However, the molecular mechanisms of ferroptosis in the context of PNI and nerve regeneration remain unclear.Ferroportin (Fpn), the only known mammalian nonheme iron export protein,plays a pivotal part in inhibiting ferroptosis by maintaining intracellular iron homeostasis.Here,we explored in vitro and in vivo the involvement of Fpn in neuronal ferroptosis.We first delineated that reactive oxygen species at the injury site induces neuronal ferroptosis by increasing intracellular iron via accelerated UBA52-driven ubiquitination and degradation of Fpn, and stimulation of lipid peroxidation.Early administration of the potent arterial vasodilator,hydralazine(HYD),decreases the ubiquitination of Fpn after PNI by binding to UBA52, leading to suppression of neuronal cell death and significant acceleration of axon regeneration and motor function recovery.HYD targeting of ferroptosis is a promising strategy for clinical management of PNI.
1.Introduction
Each year, millions of individuals worldwide experience peripheral nerve injury(PNI),which leads to long-term disability and high medical costs,resulting in negative socio-economic effects[1].PNI is usually caused by lacerations,contusions,and tractions that can arise from traumatic injuries, drug injection injuries, electrocution, infections, and disorders of metabolism [2].The peripheral nervous system possesses a certain ability to regenerate, enabling axonal regrowth after axon damage [3].Several studies have shown that treatment with natural medicinal compounds, drugs, and gene manipulation strategies can enable the recovery of nerve functions after mild-to-moderate injury [4].However, functional recovery is substantially lower after severe nerve injury.In cases of PNI,lesions at the interface between the central and peripheral nervous systems can result in neuronal loss of 35%-40% within two months after surgery, leading to a poor prognosis [5].Therefore, development of novel neuro-therapeutic approaches will require thorough comprehension of neurodegeneration/regeneration mechanisms,especially those undeciphered mechanisms potentially involved in PNI.
Cell death is a basic biological process that is required for normal development, aging, and tissue homeostasis,and its dysregulation is observed in multiple disorders[6].Recent studies have identified ferroptosis in the pathophysiology of many diseases, which differs from other types of cell death at the levels of morphology,biochemistry, and genetics [7].In ferroptosis, iron-dependent signaling causes lipid peroxides to accumulate within cells,resulting in cell death.Ferroptosis was first observed after erastin treatment that selectively kill genetically engineered cells with an oncogenic mutation [8].Recent findings have demonstrated that ferroptosis is involved in multiple neuronal diseases [9] and its inhibition can prevent neurodegeneration [10].Nevertheless, the role of ferroptosis in the pathophysiology of PNI is yet to be elucidated.
PNI is accompanied by oxidative damage and inflammation[11].Studies have recently shown that spinal nerve injury can generate reactive oxygen species(ROS)in microglial cells of the dorsal horn[12].In addition, the inhibition of oxidative stress improves functional recovery after PNI [13].Therefore, oxidative stress is considered as a primary contributor to neuronal injury and neuron loss[14].The number of residual sensory neurons following PNI is known to depend on the level of cell death within the dorsal root ganglia cells(DRGs).An in vivo study showed that up to 40%of DRGs undergo cell death within two months after PNI, compromising functional recovery [15].So far, previous studies have largely attributed neuronal loss after PNI to apoptosis.However, as our understanding of cell death has deepened over the past years,great attention has shifted to ferroptosis, a process that is triggered by ROS.Nevertheless, despite the well-defined role of ROS in PNI,whether ferroptosis is involved in the pathophysiology of nerve injury/repair has not been elucidated.In addition, the relationship between ferroptosis and functional recovery after PNI also remains unclear.The present study aimed to answer these questions.
2.Material and methods
2.1.Ethics approval
The living conditions of all animals and all experimental procedures complied with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health; Publication No.85-23,revised 1985)and were performed after approval from the Animal Research Committee of The Fourth Military Medical University,Xi'an,China(Approval No.:IACUC-20230062).All Sprague-Dawley(SD) rats used in the present study were sourced from the same center.
2.2.Drugs
H2O2and diphenyleneiodonium (DPI) were purchased from Sigma-Aldrich (St.Louis, MO, USA); and N-acetylcysteine (NAC),hydralazine(HYD),ferrostatin-1(Fer-1),and pyridoxal isonicotinoyl hydrozone(PIH)were purchased from MedChemExpress(Shanghai,China).Stock solutions of H2O2(13 mM) and NAC (30 mM) were dissolved in saline, while those of DPI (100 μM), Fer-1 (6 μM), and PIH(1 mM)were dissolved in dimethyl sulfoxide(DMSO).
2.3.Development of sciatic nerve injury (SNI) model
Adult male SD rats (200-250 g) were housed in groups of five per cage under controlled temperature(23±2°C),humidity(35%-60%), and 12-h light/12-h dark cycle.The rat model of SNI was developed using a modified version of a published protocol [16].Rats were anesthetized with intraperitoneal (i.p.) injection of 1%pentobarbital sodium (40 mg/kg; Amresco, Radnor, PA, USA).The left sciatic nerve was exposed with a 1 cm incision at the center of the thigh.The sciatic nerve was crushed at approximately 20 mm from the L4-L6 dorsal root ganglions in the orthogonal direction(2×20 s)to induce nerve injury.The incision was closed using nondegradable sutures.The same procedure was carried out in the sham group but without any induction of injury to the sciatic nerve.
Rats were subsequently allocated to three groups at random:SNI with no treatment, immediate treatment, and delayed treatment, respectively.Treatment consisted of HYD (20 μg/kg body weight) via i.p.injections once daily for seven consecutive days,starting right after (immediate group) or one week after injury induction (delayed group).The sham and SNI with no treatment groups received equal volumes of saline via i.p.injections.The concentrations of HYD well below 0.3 mg/kg, which is lowest effective concentration to obtain antihypertensive effect.
2.4.Detection of superoxide levels
We examined superoxide levels by detecting 2-hydroxyethidine,a product of dihydroethidium(DHE;Invitrogen,Carlsbad,CA,USA)oxidation.Rats were administered with 3 mL of DHE(1 mg/mL,i.p.)2 h before and 8 h after operation,sacrificed 4 h later with sodium pentobarbital(40 mg/kg,i.p.)injection,and perfused with 0.9%saline and 200 mL of 4%paraformaldehyde(PFA;Servicebio,Wuhan,China).Tissue samples from each rat was post-fixed in 4%PFA for 6 h,dehydrated in 30%sucrose for 12 h and sectioned into 20-μm thick slices using a cryocut microtome (Leica CM3050S, Weztlar, Germany).Sections were mounted onto gelatin-coated slides with vector shield for imaging on a confocal microscope (A1+, Nikon,Tokyo, Japan).At least six fields from each section were analyzed using ImageJ(National Institutes of Health,Bethesda,MD,USA).
2.5.Isolation and culture of DRGs
DRGs were isolated from neonatal (postnatal day 0-1) SD rats using previously published protocols[17].DRGs were seeded onto rat tail collagen (Invitrogen) coated coverslips and cultured in neurobasal medium (Gibco,Grand Island,NY,USA)with 50 ng/mL nerve growth factor(R&D Systems,Minneapolis,MN,USA),2 mM Lglutamine (Gibco), 2% B27 (Gibco), and 1% penicillin-streptomycin(Procell, Wuhan, China).Fibroblasts in the cultures were eliminated with 10 mM cytosine arabinoside(Sigma-Aldrich)to obtain a pure DRGs culture.
2.6.Assessment of cell viability
DRGs were seeded in 24-well plates (3000 cells/well,n=4 per group) for 24 h, followed by H2O2treatment at the designated concentration for another 24 h.Erastin(20 μM;Sigma-Aldrich)was used as a positive control.Some cells were also treated with z-VADFMK (10 μM; MedChemExpress), Fer-1 (1 μM), or PIH (100 μM) in addition to H2O2.The LIVE/DEAD Cell Imaging Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA) was used based on the manufacturer's instructions to estimate cell viability.Live (green) and dead (red)cells were quantified by ImageJ.
2.7.Detection of intracellular ROS concentration
ROS production was examined using 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA; Beyotime; Shanghai, China).DRGs were cultivated in the DRGs culture medium for 24 h(8×104cells/well)and replaced with fresh medium containing the indicated concentrations of H2O2for a further 24 h.Subsequently,cells were incubated with 10 μM DCFH-DA for 15 min and imaged under a fluorescence microscope (Olympus, Tokyo, Japan).Fluorescence intensity was measured using ImageJ.
2.8.Assessment of lactate dehydrogenase (LDH) and glutathione(GSH) levels
We assessed the GSH and LDH concentrations using the S0055 and C0016 commercial kits from Beyotime, respectively, as described by the manufacturer.
2.9.Apoptosis assay
Apoptosis assay was conducted as published [18].Briefly, cells with and without H2O2treatment were stained with annexin-V(Beyotime) for 15 min, followed by propidium iodide (Beyotime)for 5 min at room temperature.Stained cells were visualized with flow cytometry.
2.10.Heme measurement
The QuantiChrom Heme Assay Kit (DIHM-250, BioAssay Systems, Bay Area, CA, USA) was used as described by the manufacturer to determine serum and tissue heme levels.Serum or tissue homogenates (10 μL each) were mixed with the appropriate reagents and the absorbance measured at 400 nm.Heme concentration of samples were derived from a standard curve of known heme concentrations.
2.11.Lentivirus (Lv) and plasmid transfection
Primary neurons cultured in 6-well plates with neurobasal media were transduced with Lv carrying ferroportin (Fpn) (pLVEf1a-Fpn-3Flag-Puro),vector only(pLV-Ef1a-3Flag-Puro),or short hairpin RNA (shRNA) targeting UBA52 (LV2-pGLVu6-shUBA52-puro) at an multiplicity of infection of 100.All Lv were purchased from Genechem(Shanghai,China).Stably transduced cells were selected with puromycin (50 μg/mL) after 72 h.Overexpression of Fpn was confirmed by quantitative real-time polymerase chain reaction(qPCR)and Western blot assays.All plasmid transfections were performed with Lipofectamine 2000 Transfection Reagent(Thermo Fisher Scientific Inc.)as described by the manufacturer.
2.12.Assessment of iron levels
Intracellular iron levels were estimated with FerroOrange probes (DojinDo, Kyushu Island, Japan).DRGs were cultivated in DRGs medium for 24 h (8 × 104cells/well), followed by replacement with medium containing erastin, H2O2, or HYD for another 24 h.Cells were stained with FerroOrange, imaged with a flurorescence microscope,and the color intensity assessed using ImageJ.
2.13.Assessment of lipid peroxidation
The levels of lipid peroxidation were examined using the C11-BODIPY581/591method (GlpBio, Montclair, CA, USA) and were further verified with detection of malondialdehyde (MDA, Beyotime) as instructed by the manufacturer.Cells were imaged with a fluorescence microscope and the number of green and red cells counted with ImageJ.
2.14.qPCR
Total RNA from the left DRGs (L4-L6) and sciatic nerve tissue was extracted and reverse transcribed with TRIzol (Thermo Fisher Scientific Inc.) and M-MLV Reverse Transcriptase (Thermo Fisher Scientific Inc.),respectively.qPCR was carried out on the 7500 qPCR system (Applied Biosystems) and the messenger RNA (mRNA)expression relative to β-actin was calculated with the 2-ΔΔCt method[19].Primer sequences for the evaluated genes are listed in Table S1.All qPCR analyses were conducted with no less than three independent samples.
2.15.Western blot
Tissue samples and cells were homogenized in ice-cold hypotonic lysis buffer containing proteinase inhibitors and resuspended in nuclear extract buffer containing proteinase inhibitors.Protein levels were quantified with the bicinchoninic acid protein assay kit(Beyotime).Proteins were denatured by boiling for 15 min, followed by sodium dodecyl-sulfate polyacrylamide gel electrophoresis(SDS-PAGE)separation and transfer to polyvinylidene fluoride membranes (Thermo Fisher Scientific Inc.).Membranes were blocked with 5% skim milk in tris buffered saline with tween-20(TBST; Servicebio) and incubated with the following primary antibodies at 4°C overnight: anti-SLC7A11 (ab175186; Abcam, Cambridge, UK), anti-glutathione peroxidase 4 (GPX4) (ab125066;Abcam),anti-transferrin receptor 1(TfR1,ab214039;Abcam),antiferritin H (FtH, ab183781; Abcam), anti-ferritin L (FtL, ab109373;Abcam), anti-Fpn (ab239583; Abcam), anti-GAP43 (ab16053,Abcam), and anti-β-actin (Cwbio, Taizhou, China).Subsequently,membranes were washed three times with TBST, incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h, and the bound antibodies revealed with enhanced chemiluminescent substrates (Bio-rad).Each sample was assayed in triplicate.
2.16.Mitochondrial morphology assays
For ultrastructural analysis of mitochondrial morphology, cells were prepared for transmission electron microscopy (TEM) as follow:fixation in 2.5%glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) and postfixed in 2% aqueous osmium tetroxide;gradual ethanol (30%-100%) and propylene oxide dehydration;Epon (Sigma-Aldrich) embedding at 60°C for 24 h; ultra-thin sectioning; section staining with 0.5% uranyl acetate (30 min) and 3%lead citrate(7 min)at 20°C;TEM analysis(Zeiss Libra 120 Plus,Carl Zeiss, Oberkochen, Germany); and imaging with a slow scan charge coupled device-camera and iTEM software (Olympus).
2.17.Silver staining and qualitative protein analysis
For silver staining,protein samples were separated on SDS-PAGE and processed with the Fast Silver Stain Kit (Beyotime) as instructed by the manufacturer.Qualitative protein analysis was performed by the Shanghai Bioprofile Institution (Shanghai, China).Protein samples were used for digestion, high performance liquid chromatography (HPLC), and mass spectrometry (MS) based on Bioprofile's protocol.The bioinformatics pipeline involved processing of raw data (filtering and quality control), alignment of experimental MS/MS data to published databases, and identification of proteins.
2.18.Terminal deoxynucleotidyl transferase dUTP nick-end labeling(TUNEL) assay
Percentage of cell death was determined by the TUNEL assay(Beyotime) as instructed by the manufacturer.Briefly, cells were fixed in 4%PFA,permeabilized with 0.3%Triton X-100(Servicebio)for 1 h, and incubated with the TUNEL detection solutions in the dark at 37°C for 1 h.Following nuclei staining with 4′,6-diamidino-2-phenylindole (DAPI; Servicebio), slides were coversliped and imaged with a fluorescence microscope (Olympus).
2.19.Co-immunoprecipitation (Co-IP)
Co-IP was performed as published[20].Cells were lysed with a weak radioimmunoprecipitation assay lysis buffer (Cell Signaling Technology,Danfoss,MA,USA),and the lysate pre-cleared with 50%protein A/G agarose for 1 h.2 μg of primary antibody was added to 500 mL of extracted proteins and incubated overnight at 4°C.Protein A/G agarose was added to pull down the immune complexes in a 4°C shaker for 4 h.Protein were eluted from the A/G beads through boiling in 1× loading buffer and used for immunoblotting analysis.
2.20.Immunofluorescence staining
Four weeks post-surgery, regenerated nerve segments were obtained from rat models of SNI,and sectioned longitudinally after paraffin embedding.The sections were incubated with the rabbit anti-GAP43 (1:400; ab16053, Abcam) primary antibody, followed by the goat anti-rabbit IgG cy3(1:200;Abcam)secondary antibody.Each incubation was preceded by a 5 min phosphate-buffered solution wash, and all experiments were repeated thrice.
2.21.Functional assays
Sciatic function index (SFI): to evaluate behavioral outcomes after nerve injury and repair,we tracked the gait of rats 1-4 weeks after surgery.Rats were allowed to walk inside a narrow passage(5.0 cm × 8.0 cm × 45.0 cm) with a white paper-lined floor and a dark goal box at one end.Rat paws were painted with a thin layer of acrylic paint to visualize and record their footprints as they walked down the track.The SFI was calculated as follows:
In this formula, PL stands for print length and means the distance between the 3rd toe tip and the heel;TS stands for toe spread and means the distance between the 1st toe and 5th toe; and IT stands for intermediary toe spread and means the distance from the 2nd toe to the 4th toe.In addition, E means the distance measured from the operation-side of the paw, and N means the distance measured from the contralateral side of the paw.For example,EPLmeans the PL measured from the operation-side of the paw.SFI values could range from 0 (good recovery) to -100 (total dysfunction).
2.21.1.Target muscle histology
The operated hind limb was harvested four weeks post-surgery,and the gastrocnemius muscles extracted for weighing and normalization to the muscle on the unoperated side.Transverse sections (10 mm thick) of the isolated muscles were stained with the Masson trichrome stain.Six random fields were imaged across the muscle fiber sections under brightfield microscopy and the images analyzed with ImageJ.
2.21.2.Electrophysiological assays
Studies were performed at four weeks post-surgery as detailed in earlier studies[21].Rats were anesthetized with isoflurane(RWD Life Science Co., Ltd., Shenzhen, China) and the exposed sciatic nerve was clamped between two electrode placed 2 cm apart.Compound muscle action potentials (CMAP) from the gastrocnemius belly on the ipsilateral side were recorded using a multichannel electrophysiological recorder.The peak amplitude of the CMAP, nerve conduction velocity, and the latency of CMAP onset were quantified and compared across groups.
2.22.Statistical analysis
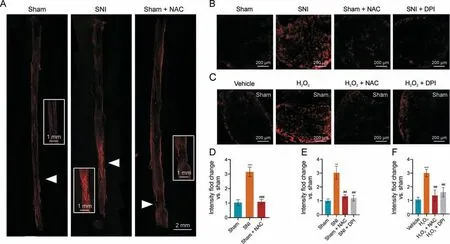
Fig.1.Sciatic nerve injury increases superoxide production.(A,B)Representative fluorescence images of dihydroethidium(DHE)staining of superoxide in sciatic nerves 2 h(A)and 12 h(B)following sciatic nerve injury(SNI),with and without N-acetylcysteine(NAC)treatment.(C)Representative fluorescence images of DHE staining in dorsal root ganglia cells(DRGs)12 h after H2O2 treatment.(D-F) Quantification of DHE fluorescence for experiments in Figs.1A-C, respectively (n = 6 per group).**P <0.01 and ***P <0.001 vs.sham or vehicle; ##P <0.01, and ###P <0.001 vs.SNI or H2O2 treatment.DPI: diphenyleneiodonium.
All statistical analyses were performed using GraphPad Prism(Version 8.0,GraphPad)and the data expressed as mean±standard error of the mean.Data were analyzed and compared using oneway analysis of variance, with Tukey's post hoc tests.P <0.05 was designated as the threshold for statistical significance.
3.Results
3.1.ROS increase at the injury site and in DRG cell bodies after PNI
Since ROS and redox stress are major contributors to cell death,we speculated that this may also occur in damaged DRG neurons and leads to neuronal cell death.Thus, we first evaluated ROS production and redox stress by detecting the expression of oxidized DHE in ex vivo DRG and sciatic nerve cultures.DHE fluorescence was significantly increased around the injury site as early as 2 h following SNI (Figs.1A and D) and could be detected in DRG 12 h after injury (Figs.1B and E), indicating that ROS levels in DRG neurons significantly increased after SNI.This injury-induced increase in ROS can be blocked by the oxidant scavenger,NAC,in both the sciatic nerve (Figs.1A and D) and the DRG (Figs.1B and E).In addition, injury-induced redox stress can also be demonstrated with H2O2treatment of uninjured sciatic nerve, which similarly increased DHE fluorescence and can be blocked by NAC (Figs.1C and F).Further supports for these findings can be seen in increased staining with the pan-ROS probe, hydrocyanine-Cy3, in SNI samples(Fig.S1),and the non-selective inhibitor of ROS-producing flavoenzymes, DPI, was as effective as NAC at reducing hydrocyanine-Cy3 staining.
3.2.Ferroptosis is an important type of cell death in DRGs after H2O2 treatment
Several methods have been reported to induce DRGs damage ranging from mechanical stimulation [22] to chemical stimulation[23].Our results showed that the level of ROS mounted up at the injury site, especially at DRG cell body after PNI.Compared with hypoxia and gamma rays,H2O2was applied in our study to trigger cellular oxidative stress in DRGs for its good stability, practical basis, and wide application in previous reports.In addition, H2O2treatment produced similar oxidative responses as SNI in vivo.Therefore, we established a model of oxidative stress using H2O2in vitro for further studies.First, DRGs were treated with various concentrations of H2O2to investigate the relationship between H2O2treatment and ROS generation.H2O2treatment increased ROS production in DRGs,as shown by increased DCFH-DA fluorescence intensity, in a dose-dependent manner (Figs.2A and C), and dramatically increased cell death (40.3% in DRGs) at the 1 mM concentration (Figs.2B and D), suggesting that oxidative injury occurred in DRGs after H2O2treatment.To determine the type of cell death that occur following H2O2treatment,we found that H2O2in the millimole range substantially increased apoptosis (Fig.S2A)and autophagy(Figs.S2B-D)in DRGs.Interestingly,H2O2-induced cell death was partially, but not completely, attenuated by z-VADFMK,an inhibitor of apoptosis(Figs.2B and D).This suggested that the mode of H2O2-induced cell death was not limited to apoptosis,and other forms of cell death, such as ferroptosis, may also be involved.
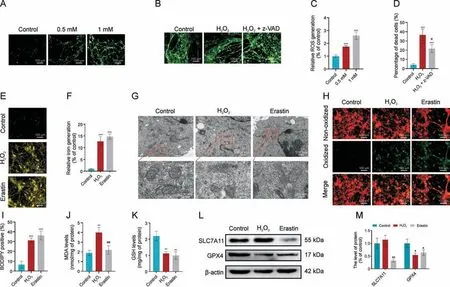
Fig.2.H2O2-induced cell death is associated with ferroptosis.(A, C) Images (A) and quantification (C) of 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) staining of reactive oxygen species (ROS) in dorsal root ganglia cells (DRGs) exposed to different concentration of H2O2.(B, D) Images (B) and quantification (D) of the percentage of dead DRGs (red)with H2O2 (1 mM) or H2O2 + z-VAD-FMK (10 μM) treatment.(E, F) DRGs treated with H2O2 (1 mM) or erastin (20 μM) were subjected to imaging (E) and quantification (F) of FerroOrange staining of intracellular iron levels.(G) Transmission electron microscopy (TEM) analysis of mitochondrial ultrastructure.(H, I) Imaging (H) and quantification (I) of C11-BODIPY581/591 staining, with green fluorescence indicating lipid peroxidation.(J) Malondialdehyde (MDA) levels determination.(K) Glutathione (GSH) content determination.(L, M) Western blot (L) and quantification (M) of SLC7A11 and glutathione peroxidase 4 (GPX4) protein levels.*P <0.05, **P <0.01, and ***P <0.001 vs.control; #P <0.05 and##P <0.01 vs.H2O2 treatment.
To test the possible involvement of ferroptosis, we first measured the changes in intracellular iron levels in DRG cultures after H2O2treatment and observed a 12.71-fold increase(Figs.2E and F).Morphologically, H2O2-treated cells displayed shrunken mitochondria,ruptured outer mitochondrial membrane,and light vacuoles that are likely related to the collapsed mitochondria(Fig.2G).These features are characteristic of ferroptosis [24], and comparable to those induced by erastin treatment (Fig.2G), a known ferroptosis inducer.In addition, H2O2treatment also elevated the production of membrane lipid peroxides(Figs.2H-J).It has been well established that ferroptosis is typically dependent on reduced expression of GPX4 and GSH, leading to excess lipid peroxidation [25].We observed that both H2O2and erastin treatment significantly reduced GSH levels (Fig.2K) and GPX4 expression (Figs.2L and M) in DRGs.We next examined the expression of SLC7A11, which transports the GSH precursor cysteine into the cytosol [26], and found that it was significantly inhibited by erastin but not by H2O2(Figs.2L and M).Together,these results indicate that ferroptosis is involved in H2O2-induced cell death in DRGs.
3.3.Inhibition of ferroptosis increases the number of DRGs after H2O2 treatment
To confirm that ferroptosis does indeed participate in H2O2-induced cell death,we performed studies with PIH,an iron chelator,and Fer-1, a strong selective inhibitor of ferroptosis.Both PIH and Fer-1 treatment significantly reduced H2O2-induced cell death and LDH levels (Figs.3A-C).H2O2-induced increase in lipid peroxidation and MDA levels were also reduced by PIH and Fer-1 (Figs.3D-F).Further studies showed that PIH and Fer-1 significantly increased GPX4 expression in H2O2-treated cell cultures (Figs.3G and H).The H2O2-induced morphological changes in mitochondria were also inhibited by PIH and Fer-1 (Fig.3I), indicating that ferroptosis does contribute to H2O2-induced cell death.
3.4.Fpn is associated with ferroptosis in DRGs
ROS may induce ferroptosis by degrading heme to release free iron or regulate the transportation of free iron.Evaluating possible effects on heme levels,we found that SNI did not alter the levels of heme in the DRG, sciatic nerve, liver,spleen,and serum compared with sham surgery (Figs.4A and B).Hemoglobin levels were also unaltered between the SNI and sham groups (Fig.4C).We then evaluated the expression of iron-related genes in DRG, including TfR1,FtH,FtL,and Fpn,and found no significant difference between the SNI and sham groups at the mRNA level(Fig.4D).Interestingly,the level of Fpn protein,the only known mammalian nonheme iron export protein[27],was reduced in the DRG after SNI(Figs.4E and F), suggesting that ROS-related ferroptosis may involve Fpn.To verify this,we found that H2O2administration caused aggregation of free iron in the DRGs, which can be suppressed by Fpn overexpression (Figs.4G and I).TUNEL assay indicated that Fpn overexpression can inhibit H2O2-induced DRG damage(Figs.4H and J),while Western blot showed that the H2O2-induced reduction in GPX4 level can be rescued by Fpn overexpression (Figs.4K and L).These data support the involvement of Fpn in H2O2-mediated ferroptosis and warrants further exploration.
3.5.Ferroptosis is induced by Fpn ubiquitination and degradation in DRGs
Although the mRNA expression of Fpn did not change in the sciatic nerve and DRG following SNI (Fig.4D), significant decrease in Fpn proteins was observed(Figs.4E and F),suggesting that redox regulation of Fpn protein may be required for neuronal ferroptosis.Thus, we next investigated possible mechanisms by which H2O2may regulate Fpn in vitro,such as via protein degradation pathways.Pretreatment of DRGs with the protein synthesis inhibitor, cycloheximide(CHX),reduced Fpn protein levels over time,and addition of H2O2enhanced Fpn degradation(Figs.5A-C).Next,we explored the two major protein degradation mechanisms, the autophagylysosomal pathway and ubiquitin-proteasomal pathway, with bafilomycin A1 (lysosome inhibitor) and bortezomib (proteasome inhibitor) treatments, respectively.The H2O2-induced Fpn degradation can be blocked by bortezomib alone or combined with bafilomycin A1, but not by bafilomycin A1 alone (Figs.5D and E),suggesting that H2O2primarily degrades Fpn via the ubiquitinproteasomal pathway.
To confirm that Fpn can interact with ubiquitin, DRGs were cotransfected with plasmids expressing Flag-Fpn and HA-ubiquitin,and whole-cell lysates were used for Co-IP with anti-Flag or anti-HA antibody conjugated agarose beads.The Co-IP data showed that Fpn does indeed interact with ubiquitin with anti-HA, anti-Fpn, and anti-Flag antibodies, all able to detect ubiquitinated species of Fpn in the DRG lysates (Figs.5F-H).Next, using the proteosome inhibitor,Z-Leu-Leu-Leu-al(MG132),to enhance ubiquitin accumulation, we found increased ubiquitination of proteins in H2O2-treated DRGs (Fig.5I).Co-IP with anti-Fpn showed that ubiquitination of Fpn is also increased by MG132 treatment,but the effect of H2O2is greater, while combined treatment with MG132 and H2O2synergistically enhanced Fpn ubiquitination(Figs.5J and K), suggesting polyubiquitination of Fpn.This interaction is likely specific since Co-IP with anti-IgG antibody did not reveal specific bands.Altogether,these results indicate that H2O2may reduce Fpn by enhancing the ubiquitination of this protein and directing it toward proteasomal degradation.
3.6.HYD inhibits Fpn degradation in DRGs by suppressing UBA52 binding to Fpn
To identify the specific ubiquitin protein that regulate Fpn ubiquitination and degradation, proteins from neuronal lysates were pulled down with the Fpn antibody for silver staining and liquid chromatography(LC)-MS,which identified a single ubiquitin protein, UBA52 (Figs.6A and B).UBA52 co-localized with Fpn in DRGs (Fig.6C), pulled-down with Fpn, and is increased with H2O2treatment along with overall ubiquitination, while Fpn level decreases (Fig.6D).To further explore the function of UBA52, we knocked down UBA52 with shRNA and found that the morphological changes induced by H2O2in primary neurons, including thinning and fragmentation of the neurites, formation of large vacuoles and bright spots, as well as decreased cytoplasm, can be rescued by shUBA52 (Fig.S3A).Ultrastructural characteristics of ferroptosis, such as the altered mitochondrial structure in H2O2-treated cells, was also inhibited by shUBA52 administration (Fig.S3B).UBA52 can interact with different signaling proteins via different functional domains.To determine which domain of UBA52 interacts with Fpn, we generated three Myc-tagged UBA52 domains (amino acids (aa) 41-117, aa 120-167, and the whole aa 1-169;Fig.6E,top panel)to perform Co-IP with Fpn.Only domain(aa 41-117) showed specific interaction with Fpn (Fig.6E bottom panel).Interestingly, the UBA52 domain (aa 41-117) is conserved across species (Fig.S4).
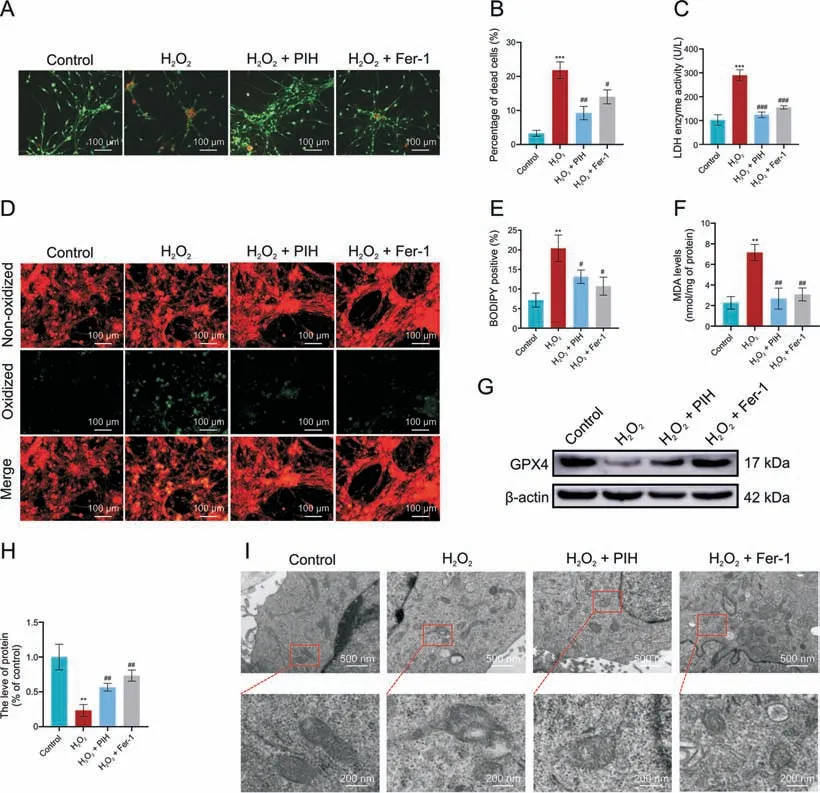
Fig.3.Iron chelation and ferrostatin-1(Fer-1)treatment inhibit H2O2-induced ferroptosis.(A,B)Dorsal root ganglia cells(DRGs)treated with H2O2(1 mM)alone or with pyridoxal isonicotinoyl hydrozone (PIH) (1 mM) or Fer-1 (6 μM) for 24 h were subjected to imaging (A) and quantification (B) of dead DRGs (red).(C) Lactate dehydrogenase (LDH) level determination.(D, E) Imaging (D) and quantification (E) of C11-BODIPY581/591 staining, with green fluorescence indicating lipid peroxidation.(F) Malondialdehyde (MDA) level determination.(G, H) Western blot (G) and quantification (H) of glutathione peroxidase 4 (GPX4) protein expression.(I) Transmission electron microscopy (TEM) analysis of mitochondrial morphology.**P <0.01 and ***P <0.001 vs.control; #P <0.05, ##P <0.01, and ###P <0.001 vs.H2O2 treatment.
Since degradation of Fpn may contribute to neuronal death after injury, a potential therapeutic strategy is to reduce/prevent Fpn degradation, and disrupting UBA52-Fpn binding may achieve this.Thus, we interrogated the Drug Signatures Database (DSigDB),which contains detailed information on existing drugs,their targets and activity data,to find compounds that may disrupt the UBA52-Fpn binding.Based on the structure of UBA52, nine compounds were predicted,of which two,HYD and deltamethrin,were further analyzed.Utilizing the Protein Data Bank database, we evaluated the predicted protein structure of the UBA52 and calculated the energy required for interaction with UBA52.Both HYD and deltamethrin possessed sufficiently high binding energies to interact with UBA52(Figs.6G and H).However,deltamethrin is a common insecticide that has demonstrated neurological,cardiovascular,and reproductive toxicity in various aquatic organisms [28].Therefore,HYD was selected for subsequent biological testing.Conjugating HYD to biotin allowed us to perform an affinity pull-down assay(Fig.6I) and showed that HYD-biotin can bind to endogenous UBA52 in DRGs (Fig.6J).To examine whether HYD affect H2O2-induced Fpn degradation, we performed Co-IP assays after treatment with HYD.HYD treatment increased Fpn levels with or without H2O2stimulation and reduced the binding of UBA52 and ubiquitin to Fpn.However, HYD had little effect on UBA52 levels(Fig.6K).
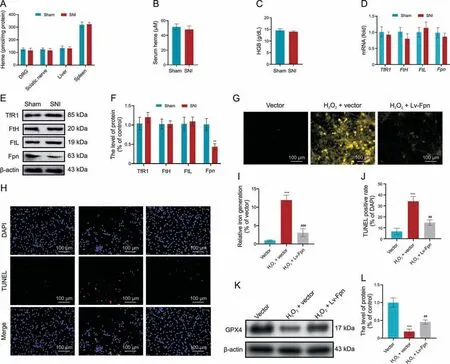
Fig.4.Sciatic nerve injury(SNI)promotes rapid and systemic iron overload independent of heme degradation.(A,B)Four days after SNI,heme concentrations were determined for various tissues (A) and serum (B).(C) Hemoglobin (HGB) levels were measured.(D-F) Expression of iron-related genes (D) and proteins (E, F) were measured.(G, I) Dorsal root ganglia cells(DRGs)transduced with lentivirus(Lv)overexpressing ferroportin(Fpn)and treated with H2O2 were subjected to imaging(G)and quantification(I)of intracellular iron levels.(H, J) imaging (H) and quantification (J) of terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining.(K, L) Western blot(K) and quantification (L) of glutathione peroxidase 4(GPX4)protein levels.**P <0.01 and ***P <0.001 vs.sham or vector; ##P <0.01 and ###P <0.001 vs.H2O2+vector treatment.TfR1:transferrin receptor 1;FtH: ferritin H; FtL: ferritin L; DAPI: 4′,6-diamidino-2-phenylindole.
As HYD can reverse the H2O2-induced Fpn degradation, it is likely to inhibit ferroptosis in H2O2-treated cells.To confirm this,DRGs were treated with H2O2or H2O2+HYD for 24 h and iron level,cell death, lipid peroxidation, and GPX4 levels were evaluated.H2O2-induced increases in iron production (Figs.7A and B), cell death (Figs.7C and D),and lipid peroxidation (Figs.7E and F), and decrease in GPX4 expression(Figs.7G and H)were all rescued with HYD treatment.Findings of the in vivo studies are also consistent with the above in vitro data.Significantly higher levels of iron were observed in the SNI group compared to the sham group, and HYD can reduce this iron accumulation in DRGs after SNI (Fig.S5A).In addition,Western blot and IHC results indicated that HYD can also rescue Fpn expression after SNI (Figs.S5B and C).These results revealed that HYD treatment significantly inhibits DRGs ferroptosis induced by ROS.
3.7.HYD increases sciatic nerve regeneration by blocking ferroptosis
The ability of HYD to rescue Fpn expression in vitro and in vivo suggest that it may have therapeutic potential.To verify this, HYD was administered to rats immediately or one week (delayed administration)after SNI(Fig.8A),followed by evaluation of axonal regeneration four weeks after injury.The length(from crush site)of axons positive for GAP43, a marker of axonal regeneration, was longest in the immediate HYD treatment group, followed by the delayed HYD treatment group and the untreated control group;differences among all groups were significant (Fig.8B), and Western blot quantifications confirmed these results (Figs.8C and D).These results demonstrate that inhibiting ferroptosis improves axonal regeneration following SNI.
Gait is one of the primary indexes for evaluating sciatic nerve function and recovery.At two weeks after SNI, no significant difference was found between rats with and without HYD treatment,indicating a lack of functional recovery at this time-point across all groups.However, by four weeks after SNI, rats who had received HYD treatment showed significantly fuller footprints while walking(Fig.8E) and higher SFI values (Fig.8F), indicating that the inhibition of ferroptosis can promote functional neurological recovery following SNI.In addition, immediate HYD treatment produced better outcomes than delayed HYD treatment (Figs.8E and F),suggesting that inhibiting ferroptosis immediately after injury is more effective in promoting neurological recovery.
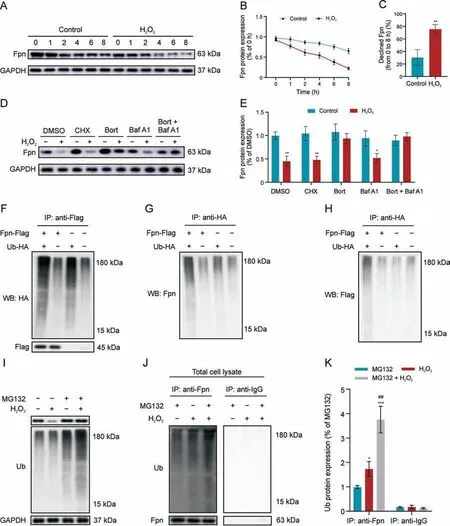
Fig.5.H2O2 promotes ubiquitination-based degradation of ferroportin(Fpn).(A-C)Dorsal root ganglia cells(DRGs)pretreated with cycloheximide(CHX)(5 μg/mL)for 24 h were stimulated with H2O2 for different length of time and subject to Western blot analysis of Fpn expression (A), quantification of Fpn protein levels over time (B), and calculation of overall changes in Fpn expression (C).(D, E) Western blot (D) and quantification (E) of Fpn protein levels after treatment with CHX (5 μg/mL), bortezomib (Bort; 200 nM), bafilomycin A1(Baf A1;50 nM),or a combination of Bort and Baf A1 for 1 h before the introduction of 1 mM H2O2.(F-H)Co-immunoprecipitation(Co-IP)of Fpn-Flag and ubiquitin-HA with Western blot analysis of HA(F),Fpn(G),and Flag(H),depicting ubiquitinated species of Fpn.DRGs were exposed to Z-Leu-Leu-Leu-al(MG132;20 μM)for 6 h prior to exposure to H2O2(1 mM)for 24 h.(I) Total lysates were analyzed for ubiquitinated proteins via immunoblotting.(J) Co-IP with Fpn or IgG antibody and Western blot with anti-ubiquitin in MG132-treated and untreated cells.(K)Quantification of Western blot images from Fig.5J.*P <0.05, **P <0.01,and ***P <0.001 vs.control or MG132; ##P <0.01 vs.H2O2 treatment.GAPDH: glyceraldehyde-3-phosphate dehydrogenase; DMSO: dissolved in dimethyl sulfoxide; Ub-HA: HA labeled ubiquitin.
The degree of gastrocnemius atrophy is also widely used to estimate the recovery of sciatic function.Masson trichrome staining of the gastrocnemius (Fig.8G) at four weeks after injury showed that HYD treatment significantly increased the average muscle fiber area(Fig.8H)and the relative wet weight ratio of the gastrocnemius (Fig.8I), with the effect of immediate treatment superior to delayed treatment.Hence, immediate inhibition of post-injury ferroptosis by HYD appeared to reduce target muscle atrophy.

Fig.6.UBA52 is crucial for H2O2-induced ferroportin(Fpn)ubiquitination.(A)Dorsal root ganglia(DRG)cell lysates immunoprecipitated with Fpn antibody and treated with silver staining to reveal all proteins bound to Fpn antibody.(B)Mass spectrogram of UBA52.(C)Immunofluorescence staining showing co-localization of UBA52(green)and Fpn(red)in primary cortical neurons.(D) Immunoprecipitation (IP) with Fpn followed by Western blot with anti-ubiquitin, anti-Fpn and anti-UBA52 in dorsal root ganglia cells (DRGs).(E)Schematic representation of UBA52 fusion proteins (top panel).Co-IP showing interaction between UBA52 domain (amino acides (aa) 41-117) and Fpn (bottom panel).(F)Schematic of the experiment that predicts the docking with small molecule drugs.(G,H)Predication of binding between UBA52 and hydralazine(HYD)(G)or deltamethrin(H)as determined by molecular docking.(I) Schematic of the experiment that detects the binding of HYD-biotin to UBA52.(J) Pull-down assay to detect the binding of HYD-biotin to UBA52 and immunoblotting analysis of the input proteins.(K)IP with Fpn followed by Western blot with anti-ubiquitin shows that the addition of HYD reduces ubiquitination of Fpn in the presence or absence of H2O2.M:marker;DSigDB:Drug Signatures Database;GAPDH:glyceraldehyde-3-phosphate dehydrogenase;Myc:protein tag(protein sequence:EQKLISEEDL); IB: immunoblotting; SDS-PAGE: sodium dodecyl-sulfate polyacrylamide gel electrophoresis; Ub: ubiquitin.

Fig.7.Hydralazine(HYD)inhibits H2O2-induced dorsal root ganglia cells(DRGs)death.(A,B)DRGs treated with H2O2(1 mM)with and without HYD for 24 h were subjected to imaging(A)and quantification(B)of intracellular iron levels.(C,D)imaging(C)and quantification (D) of dead cells (red).(E, F) Imaging (E) and quantification (F) of C11-BODIPY581/591 staining, with green fluorescence indicating lipid peroxidation.(G, H)Western blot (G) and quantification (H) of glutathione peroxidase 4 (GPX4) level.**P <0.01 and ***P <0.001 vs.control; #P <0.05 and ##P <0.01 vs.H2O2 treatment.
We also evaluated functional recovery with electrophysiology.HYD treatment significantly increased the peak CMAP amplitude(Fig.8J) and nerve conduction velocity (Fig.8K), and reduced the CMAP latency (Fig.8L).In all cases, immediate HYD treatment resulted in better outcomes than delayed treatment.These data suggest that the inhibition of ferroptosis can promote functional motor recovery after SNI, with a more profound effect obtained with immediate HYD treatment.
4.Discussion
Excess levels of ROS can cause lipid, protein, and DNA damage,augmenting disease pathogenesis [29].Cell and organelle membranes are particularly vulnerable to ROS damage because they contain high levels of polyunsaturated fatty acids.In addition,peroxidation of membrane lipids not only damages cell components but also causes structural damage, finally resulting in cell rupture and death.In previous studies,Cox4i2 triggers an increase in reactive oxygen species, leading to ferroptosis and apoptosis in HHV7infected schwann cells, which is a glia cell in the peripheral nerves [30].Furthermore, knockdown of the lysosomal protein prosaposin strongly sensitizes neurons to oxidative stress by triggering the formation of lipofuscin, which traps iron, generating reactive oxygen species and triggering ferroptosis in central nervous system [31].In our study, ROS levels were found to be increased in both the sciatic nerve and DRG following SNI, in line with earlier reports[23].
Previous studies have demonstrated that ROS production after spinal nerve transection mostly occur in the mitochondria of damaged neurons [32].Other studies have found that ROS is notably produced by macrophages and transported to the cell body via axons [33].Administration of H2O2onto the sciatic nerve significantly increases ROS in DRG.Hence,we speculated that ROS is produced by cells at the site of injury and delivered to the cell body by the axon.This conclusion is supported by the observation that administration of NAC at the injured site of the sciatic nerve can inhibit ROS production in DRG.Under normal conditions,GPX4 rapidly reduces lipid peroxides via GSH oxidation.However, GSH down-regulation owing to excess ROS accumulation leads to an increase in lipid peroxides, triggering cell death.In addition, we found that ROS increases iron accumulation, which can in turn damage the lipid and mitochondrial membranes.
Iron is an essential cofactor for many cellular enzymes involved in myelin synthesis.Thus, imbalance in iron homeostasis will contribute to peripheral neuropathies.Recent studies showed that the accumulation of iron can increase ROS levels [34], and the expression of Fpn, which is involved in maintaining iron homeostasis,is decreased in the peripheral nerve system after SNI[35].In our study,SNI significantly increased the ROS level in DRGs,and the decreased Fpn expressions is associated with increased iron in the DRGs.However, the expression of Fpn was significantly decreased after H2O2treatment, indicating that ROS was unlikely to be induced by the accumulation of iron after SNI.
We found that autophagy was significantly increased after H2O2treatment.As the autophagy-lysosomal pathway is one of the major mechanisms of protein degradation, we thus evaluated whether this pathway is involved in the degradation of Fpn after H2O2treatment.However, our data showed that ROS did not use the autophagy-lysosomal pathway for Fpn degradation.The other major protein degradation pathway involving proteosomes has been shown to degrade several plasma membrane metal transporters,including DMT1,SLC39A14,and SLC30A1[36].Fpn has also been shown to be degraded by this pathway following hepcidininduced endocytosis and ubiquitination [37].Similarly, we provide evidence in this study that ROS induced Fpn degradation principally via the ubiquitin-proteasomal pathway.
The ubiquitin-proteasome system is essential in regulating mitochondrial physiology and controls the expression of different intramembrane space and inner mitochondria membrane proteins,such as succinate dehydrogenase subunit A, and other tricarboxylic acid-cycle metabolites[38].Recently,we reported that the ubiquitin protein, UBA52, facilitates the ubiquitination of chaperone HSP90 together with the E2 and E3 ligases, leading to inhibition of endoplasmic reticulum stress and Parkinson's diseasespecific pathological markers, such as tyrosine hydroxylase and αsynuclein[39].Our current study implicates UBA52 in SNI-induced ferroptosis as inhibition of UBA52 suppressed ferroptosis after SNI.We provided evidence of direct interaction between UBA52 and Fpn following H2O2treatment, indicating that UBA52 can effectively promote Fpn degradation after H2O2administration.This prompted us to explore potential targeting drugs to this pathway to mitigate SNI.
Protein crystal structures not only highlights the potential mechanisms of action of specific proteins, but also offer insights into drugs and molecules that can modulate protein activity.Through such structural analysis of UBA52,we identified HYD and deltamethrin as potential molecules that can regulate the UBA52-Fpn interaction.Since deltamethrin is potentially toxic, we evaluated the function of HYD and found that it indeed has the potential to regulate Fpn ubiquitination by UBA52.Previous, HYD has been shown to support neuronal regeneration in a mouse model of spinal cord injury [40].However, it has not been demonstrated in protein degradation and peripheral nerve injury.Our results suggested that HYD can reduce Fpn degradation in DRG following injury, thus preventing accumulation of iron and subsequent ferroptosis.
Recovery after PNI not only relies on enhanced axonal regeneration but also on reduced cell death, including reduced ferroptosis.Our study showed that HYD not only reduces sciatic nerve necrosis but also improves functional recovery following SNI.Previous studies found that primary sensory neurons started dying within 24 h of sciatic nerve transection[41],but neuronal loss only starts between 4 and 10 days following injury [42].This gap between the onset of neuronal death and neuronal loss reflects the time taken for neurons to move from the DNA fragmentation stage to complete involution.This gap may also be the key stage for preventing cell death in neurons.Here,we showed that immediate treatment with HYD following injury were far more effective in promoting axonal regeneration and functional recovery than delayed HYD treatment, suggesting that the early inhibition of ferroptosis is vital for axonal regeneration since ferroptosis begins 0-7 days following SNI.
As a specific form of regulated cell death, ferroptosis plays crucial roles in multiple pathological conditions including ischemic organ injuries, cancer, and degenerative diseases [43];increasing evidence also implicates potential physiological functions of ferroptosis [44].Numerous organ injuries and degenerative pathologies are driven by ferroptosis [45].Multiple studies have established a role of ferroptosis in neuronal demise,including stroke and other brain injuries.An ex vivo study using rat hippocampal slice culture showed that glutamate-induced neuronal excitotoxic cell death can be blocked by radicaltrapping antioxidant ferrostatin 1 [45].As such, pharmacological modulation of ferroptosis, via both its induction and its inhibition, holds great potential for the treatment of drug-resistant organ injuries, cancers, and other degenerative diseases linked to extensive lipid peroxidation.Here, we demonstrated occurrence of ferroptosis in SNI arising from ROS-accelerated UBA52 ubiquitination and degradation of Fpn, leading to accumulation of intracellular iron and stimulation of lipid peroxidation.These findings suggest that UBA52 is a potential therapeutic target for peripheral nerve injury.Furthermore, we provided preliminarily evaluation of HYD, an U.S.Food and Drug Administrationapproved anti-hypertensive agent, as a potential treatment compound to inhibit ferroptosis in DRG by targeting UBA52.Clinical translation of HYD for restoration of peripheral nerve injury still requires further exploration and verification.Our study demonstrated a promising strategy of screening approved drug libraries for new indications.
5.Conclusion
In this study, ROS levels were found to be increased in both the sciatic nerve and DRG following SNI.Under normal conditions, GPX4 rapidly reduces lipid peroxides via GSH oxidation.However, GSH down-regulation owing to excess ROS accumulation leads to an increase in lipid peroxides,triggering ferroptosis.In addition, we found that ROS increases iron accumulation,which can in turn damage the lipid and mitochondrial membranes.In addition, the expression of Fpn was significantly decreased after H2O2treatment via the ubiquitin-proteasomal pathway.Furthermore, we found UBA52 can effectively promote Fpn degradation after H2O2administration.This prompted us to explore potential targeting drugs to this pathway to mitigate SNI.
We found HYD can reduce Fpn degradation by targeting UBA52 in DRG following injury,thus preventing accumulation of iron and subsequent ferroptosis.Recovery after PNI not only relies on enhanced axonal regeneration but also on reduced cell death,including reduced ferroptosis.Our study showed that HYD not only reduces sciatic nerve necrosis but also improves functional recovery following SNI.In addition, we showed that immediate treatment with HYD following injury were far more effective in promoting axonal regeneration and functional recovery than delayed HYD treatment, suggesting that the early inhibition of ferroptosis is vital for axonal regeneration since ferroptosis begins 0-7 days following SNI.
CRediT author statement
Shengyou Li:Conceptualization,Data curation,Formal analysis,Investigation, Writing - Original draft preparation;Xue Gao:Data curation, Formal analysis, Writing - Original draft preparation;Yi Zheng:Data curation,Formal analysis,Validation;Yujie Yang:Data curation, Investigation;Jianbo Gao:Conceptualization, Supervision; Methodology;Dan GengandLingli Guo:Data curation,Formal analysis;Teng Ma:Data curation, Funding acquisition,Formal analysis;Yiming Hao:Conceptualization, Methodology;Bin Wei:Supervision, Formal analysis, Investigation;Liangliang Huang:Data curation, Funding acquisition;Yitao Wei:Formal analysis, Investigation;Bing Xia:Conceptualization, Funding acquisition, Writing - Reviewing and Editing;Zhuojing Luo:Project administration, Funding acquisition, Writing - Reviewing and Editing;Jinghui Huang:Conceptualization, Funding acquisition,Supervision, Project administration, Writing - Reviewing and Editing.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Grant Nos.: 82122043, 81972052,81902213, 82201537, and 81730065), and the China Postdoctoral Science Foundation (Grant Nos.: 2021M693946 and 2019M653967).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.08.006.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Platycodin D inhibits angiogenic vascular mimicry in NSCLC by regulating the eIF4E-mediated RNA methylome
- Identification of different degrees of processed ginger using GC-IMS combined with machine learning
- Simultaneously quantifying hundreds of acylcarnitines in multiple biological matrices within ten minutes using ultrahigh-performance liquid-chromatography and tandem mass spectrometry
- A proteomic landscape of pharmacologic perturbations for functional relevance
- Licorice-saponin A3 is a broad-spectrum inhibitor for COVID-19 by targeting viral spike and anti-inflammation
- Distinct molecular targets of ProEGCG from EGCG and superior inhibition of angiogenesis signaling pathways for treatment of endometriosis