Dynamics and genetic regulation of macronutrient concentrations during grain development in maize
2024-03-12PengchengLiShuangyiYinYunyunWangTianzeZhuXinjieZhuMinggangJiWenyeRuiHoumiaoWangChenwuXu2ZefengYang2
Pengcheng Li ,Shuangyi Yin ,Yunyun Wang ,Tianze Zhu ,Xinjie Zhu ,Minggang JiWenye RuiHoumiao WangChenwu Xu2#,Zefeng Yang2#
1 Jiangsu Key Laboratory of Crop Genetics and Physiology/Key Laboratory of Plant Functional Genomics, Ministry of Education/Jiangsu Key Laboratory of Crop Genomics and Molecular Breeding/Agricultural College, Yangzhou University, Yangzhou 225009,China
2 Jiangsu Co-Innovation Center for Modern Production Technology of Grain Crops, Yangzhou University, Yangzhou 225009, China
Abstract Nitrogen (N),phosphorus (P),and potassium (K) are essential macronutrients that are crucial not only for maize growth and development,but also for crop yield and quality. The genetic basis of macronutrient dynamics and accumulation during grain filling in maize remains largely unknown. In this study,we evaluated grain N,P,and K concentrations in 206 recombinant inbred lines generated from a cross of DH1M and T877 at six time points after pollination. We then calculated conditional phenotypic values at different time intervals to explore the dynamic characteristics of the N,P,and K concentrations. Abundant phenotypic variations were observed in the concentrations and net changes of these nutrients. Unconditional quantitative trait locus (QTL) mapping revealed 41 non-redundant QTLs,including 17,16,and 14 for the N,P,and K concentrations,respectively. Conditional QTL mapping uncovered 39 non-redundant QTLs related to net changes in the N,P,and K concentrations. By combining QTL,gene expression,co-expression analysis,and comparative genomic data,we identified 44,36,and 44 candidate genes for the N,P,and K concentrations,respectively,including GRMZM2G371058 encoding a Doftype zinc finger DNA-binding family protein,which was associated with the N concentration,and GRMZM2G113967 encoding a CBL-interacting protein kinase,which was related to the K concentration. The results deepen our understanding of the genetic factors controlling N,P,and K accumulation during maize grain development and provide valuable genes for the genetic improvement of nutrient concentrations in maize.
Keywords: maize,nutrient concentration,unconditional QTL mapping,conditional QTL mapping,dynamic trait
1.Introduction
Maize is one of the most important cereal crops in the world,and it provides food,feed,and biofuel for human and animal consumption (Duvick 2005;Ranumet al.2014;Xieet al.2022). Maize grain quality is determined by various macronutrients,such as starch,protein,oil,and minerals,that affect its nutritional value,processing properties,and end-use performance (Guet al.2015;Sharma and Carena 2016). Nitrogen (N),phosphorus (P),and potassium (K) are essential macronutrients for maize growth and development,and their accumulation in maize grains has important implications for human and animal health (Huanget al.2020;Wanget al.2021). In the past few decades,maize yields have markedly increased,whereas the concentrations of macronutrients,especially N,have followed a downward trend (Duvick 2005;Guoet al.2019). The accumulation of nutrients in kernels is a very dynamic and complex process (Renet al.2023).Understanding the dynamics and genetic regulatory mechanisms of N,P,and K accumulation in kernels during maize seed development is crucial for improving maize grain quality by mineral biofortification.
Maize kernel development is divided into the lag,grainfilling,and maturation stages (Wuet al.2022). The lag stage of growth (~0–15 days after pollination (DAP))involves diploid embryo and triploid endosperm formation,and the differentiation of various tissues and organs in the kernel. During the grain-filling stage of seed development(~12–40 DAP),starch,protein,and oil rapidly accumulate in the endosperm,and embryonic growth takes place(Domínguez and Cejudo 2014;Wuet al.2022). The final maturation stage may involve the loss of water and development of desiccation tolerance in the kernel and the completion of embryonic maturation and dormancy induction (Wuet al.2022). In plants,N,P,and K are taken up from soil by the roots,assimilated,and then mostly transferred to the leaves with subsequent remobilization into developing grains. During the grain-filling stage,plants remobilize and transport nutrients distributed throughout vegetative source organs into the seeds (Iwaiet al.2012). For example,young developing roots and leaves in the vegetative stage act as sink organs for N and amino acids. During the subsequent reproductive stage,the N accumulated in roots and shoots undergoes protein hydrolysis and remobilization to storage organs(de Banget al.2021). Higher plant tissues typically contain approximately 1.5% N,0.2% P,and 1.0% K(Hawkesfordet al.2023). The concentrations of nutrients in mature kernels are determined by their accumulation during grain development and their subsequent dilution through carbohydrate deposition (Alfoldiet al.1994). The regulation of macronutrient concentrations in maize grains is complex and influenced by multiple factors,such as genotype,environment,and management practices,and their interactions (Ciampittiet al.2013;Guoet al.2019).The heritability of N,P,and K concentrations has been reported to be rather high: 64.1 and 66.1% for N under low N (LN) and high N (HN) conditions,respectively (Liet al.2015),and 72.0 and 74.0% for P and K,respectively(Guet al.2015). A thorough understanding of the genetic factors regulating N,P,and K deposition in seeds is especially important for the breeding of special maize lines with high N,P,and K nutrient contents. Because of the dynamics and complexity of nutrient accumulation,however,few studies have addressed the genetic features related to changes in the N,P,and K nutrient contents during seed developmental processes.
Most quantitative trait locus (QTL) studies in maize have focused on agronomic,physiological,and yieldrelated traits under different nutrient conditions (Caiet al.2012a,b;Liuet al.2013;Liet al.2015). Some QTL studies focusing on the concentrations of grain N,P,and K in seeds have also been conducted (Guet al.2015;Liet al.2015). In particular,Liet al.(2015) identified nine and five QTLs for grain N content under HN and LN conditions,respectively. In addition,a comprehensive QTL analysis based on the concentrations and contents of seven minerals in grains across four environments uncovered five,two,and five QTLs for K content,P concentration,and P content,respectively (Guet al.2015). At each time point,the contents of N,P,and K nutrients,which are affected by the level at the previous point,involve a different set of genes,thereby posing a challenge to our understanding of the genetic mechanisms. An appropriate strategy to elucidate this influence is the combination of unconditional with conditional QTL mapping to detect related loci across the entire dynamic process. Unconditional QTL mapping can be used to analyze the performance of a trait at each time point and detect the cumulative effects from the beginning of ontogeny until the measured time point. By contrast,conditional QTL mapping reflects the dynamic behavior of QTL expression and can be used to estimate the net effects of QTL expressed in each time interval(Zhu 1995;Zhaoet al.2006). Molecular dissection based on unconditional and conditional QTL mapping has been conducted to investigate the dynamic behavior of complex traits,such as seed filling (Yinet al.2020),enzyme activity in peach (Desnoueset al.2016),and seed hardness in soybean (Buet al.2018). However,this approach has not yet been used to elucidate the dynamics and genetic regulation of grain macronutrient concentrations in maize.
In the present study,we evaluated the N,P,and K concentrations at six key time points during maize seed development in a recombinant inbred line (RIL) population derived from T877 and DH1M. Our objectives were: (1)to measure the changes in N,P,and K accumulation during seed development;(2) to identify unconditional and conditional QTLs for N,P,and K accumulation;and(3) to combine QTL mapping,gene expression profiling,and genetic variant analysis to identify candidate genes related to the concentrations of N,P,and K in grain.
2.Materials and methods
2.1.Plant materials,field experiments and seed nutrient concentration determination
The RIL population consisting of 206 lines was derived from a cross between the parental genotypes DH1M and T877,which clearly differed in their N,P and K nutrient concentrations during the seed developmental process.DH1M has higher N,P and K nutrient contents than T877. In 2017,the experiment was conducted at Sanya,Hainan Province,(18°23´N,109°44´E) in China with three replicates. Each of the lines contained 78 plants that were planted in a plot consisting of six rows with a row length of 3.0 m,a distance between rows of 0.5 m and 13 plants per row. To determine the sample time,the pollination dates of the individual RILs were recorded. In a previous study,we recorded seed dry weight at 14 time points after pollination (10,15,20,25,30,35,40,43,46,49,52,55,58,and 61 DAP),spanning the early filling stages to the late maturation stages. From the seed-filling curve,we found that 10,20,30,40,49 and 58 DAP were the key times for seed development,so the samples from these times were chosen to measure the concentrations of N,P and K (Yinet al.2020). At each sampling,two ears with synchronous developmental processes were selected. Fifty seeds in the middle of each ear were removed and dried at 70–80°C to constant weight after undergoing fixation at 105°C for 1 h. The seeds of two ears were mixed and milled,and finally sieved through a 1 mm mesh. The total N concentration was determined using a modified Kjeldahl digestion method (Nelson and Sommers 1973). The total P concentration was measured by an automated colorimetric method (Soon and Kalra 1995) and the total K concentration was measured by a flame photometer.
2.2.Statistical analysis of phenotypic data
The N,P and K nutrient concentrations at different time points were represented asy(t),whereyis the corresponding nutrient andtis the number of DAP. To obtain the net effect of the nutrient contents between two adjacent time points,the conditional phenotypic values of the nutrient contents were predicted in software QGAStation2.0 (http://ibi.zju.edu.cn/software/qga/). The time-dependent conditional phenotypic values were represented asy(t|t–1),which indicated the net effect of nutrient concentrations at sequential time intervals from time pointtto time pointt–1 (Zhu 1995).
The descriptive statistics of the N,P and K concentrations at six measurement times and net effects of the N,P and K concentrations in each time interval,including mean,standard deviation,maximum,minimum,skewness,kurtosis and coefficient of variation (CV),were calculated using the R software. The changes in trajectories of the N,P and K concentrations during the seed development process were fitted by a converted Logistic model,y=1–whereyis the nutrient content;tis the number of DAP;andk,aandbare parameters. These parameters were estimated using a nonlinear least-squares approach implemented in R-4.2.0(Milliken 1990). Phenotypic correlation analysis between traits was carried out using the agricolae (v1.3-5) software package.
2.3.Construction of genetic linkage maps and QTL analysis
A high-density linkage map consisting of 3,227 bin markers had been constructed for the RIL populations derived from a cross between DH1M and T877,using the MaizeSNP50 chip. The numbers of bin markers per chromosome ranged from 111 to 503,and the total genetic distance of this linkage map was 2,450 cM,with an average genetic distance between two adjacent markers of 0.76 cM (Yinet al.2020).
The observed N,P and K concentrations at the six measurement times were used for unconditional QTL mapping,and the net effect of the N,P and K concentrations in each time interval were used for conditional QTL mapping. QTL analysis was performed using composite interval mapping (CIM),with a windows size of 10 cM and a step size of 1 cM in R/qtl (v1.50)(Bromanet al.2003). To avoid the loss of QTLs with small effects,the LOD threshold was empirically set as 3.0 and the support interval (CI) of QTL was set as a 1-LOD fall-off on either side of the LOD peak. The QTL naming was based on the method of McCouch (1997).Briefly,the QTL name was constructed as follows:q+trait name+“–”+chromosome+“–”+QTL number(McCouch 1997). To compare nutrient concentrations among inbred lines with different QTLs,a hierarchical clustering method was used to cluster the 206 lines.According to the genotypes of the detected QTLs,we extracted the genotype of each inbred line in the QTL regions. The distance matrix was calculated through Euclidean distances using the “dist” function in R,and then clustered by the “hclust” function. Based on N,P and K QTLs,the RILs were divided into five clusters,and phenotypic differences between clusters were tested using the analysis of variance (ANOVA) and least significant difference (LSD) method in R.
2.4.Time course transcriptomes during maize seed development
To characterize the expression of genes in QTLs associated with nutrient concentrations during the seed developmental process,seeds of T877,DH1M and RIL147 were sampled at 10,20,30,40 and 50 DAP.Seeds in the middle of two similar ears were sampled as two biological replicates,followed by RNA isolation and RNA-seq analysis (Yinet al.2020). The differentially expressed genes (DEGs) between two parents during the seed developmental process were identified and the reads per kilobase of exon per million mapped reads(FPKM) values of individual genes at different time points were calculated using the ballgown (v2.30.0)Software package (Frazeeet al.2015). In this study,the genes with an adjustedP-value<0.05 through false discovery rate (FDR) correction (Benjamini and Hochberg 1995) and an absolute value of log2(fold change)>1 were identified as DEGs. The gene expression values across the six time points in the three lines were used to construct the co-expression networks using the WGCNA package (v1.71) in R(Langfelder and Horvath 2008). The modules were obtained with the default settings of the automatic network construction function blockwiseModules,with minor modifications (deepSplit=2,minModuleSize=30 and mergeCutHeight=0.30). The eigengene value was calculated for each module and used to test the association with nutrient concentrations.
2.5.Whole-genome re-sequencing of the two parental lines
The DNA libraries of the two parental lines were sequenced using an Illumina HiSeq 2000 machine (Liet al.2018). The filtered high-quality sequences were aligned and mapped onto the B73 reference genome (ftp://ftp.ensemblgenomes.org/pub/ plants/release-24/fasta/zea_mays/dna/) using BWA (v0.7.17) software (Li and Durbin 2009). SNP calling was performed using SAMtools(v1.14) software (Li 2011). Low-quality SNPs with a base quality value<20 and a read depth<4 were excluded. We masked the heterozygous SNPs as missing data. Finally,the SNPs with polymorphism between the two parents were retained. The identified SNPs were annotated using the ANNOVAR (version: 2019-10-24) program (Wanget al.2010).
3.Results
3.1.Unconditional and conditional phenotypic variation
To observe the changes in N,P,and K nutrient concentrations during seed development in maize,we fitted the trajectories of the changes in nutrient concentrations with a converted logistic model (Fig.1-A). The mean determination coefficients (R2) of the fitting trajectories for N,P,and K were 0.930,0.924,and 0.988,respectively,thus indicating that these fitted curves accurately reflected the changes in nutrient concentration. The concentrations of N and P were higher in T877 than in DH1M throughout the seed developmental process. The concentration of K was also higher in T877 than in DH1M at the beginning of seed development. By the middle stage of seed development,however,the concentration of K in T877 was similar to that in DH1M,and the concentration in T877 was lower than that in DH1M by the end of the process (Fig.1-A;Appendix A). Within the RIL population,the concentrations of N,P,and K differed significantly among the time points(Fig.1-A;Appendix A). Abundant phenotypic variation was also observed,with CV values ranging from 12.9 to 18.4% for N,7.8 to 20.0% for P,and 9.4 to 28.5%for K. Interestingly,the CV values for each nutrient initially decreased and then increased during the seed developmental process. To analyze the net changes in N,P,and K nutrient concentrations,we calculated conditional phenotypic values at different time intervals and plotted their distributions. As shown in Fig.1-B,mean net changes in the three nutrients decreased during seed development.During seed development,the CVs of net changes of the three nutrients,which were lower than those of the nutrient concentration,ranged from 9.5 to 15.5% for N,5.0 to 15.6% for P,and 7.0 to 25.8% for K (Appendix B).
Pearson correlation analysis (Appendices C and D) revealed that N(58) (N concentration at 58 DAP)was significantly correlated (r=0.28–0.43) with the N concentration at all other time points except for N(10). In addition,N(40) was most strongly correlated with N(58).K(58) was significantly correlated (r=0.15–0.24) with its K concentration at all time points other than K(40),and K(49) had the highest correlation with K(58). Except for P(10),the concentration of P at each time point was significantly correlated with P(58),with P(49) having the highest correlation with P(58). Among the three nutrients,the highest correlation coefficient was that of K(58) with P(58) (r=0.76). During the seed developmental process,the correlations between P and K gradually increased(r=0.33–0.76),whereas those between N and the other two nutrients gradually decreased (Appendix C).
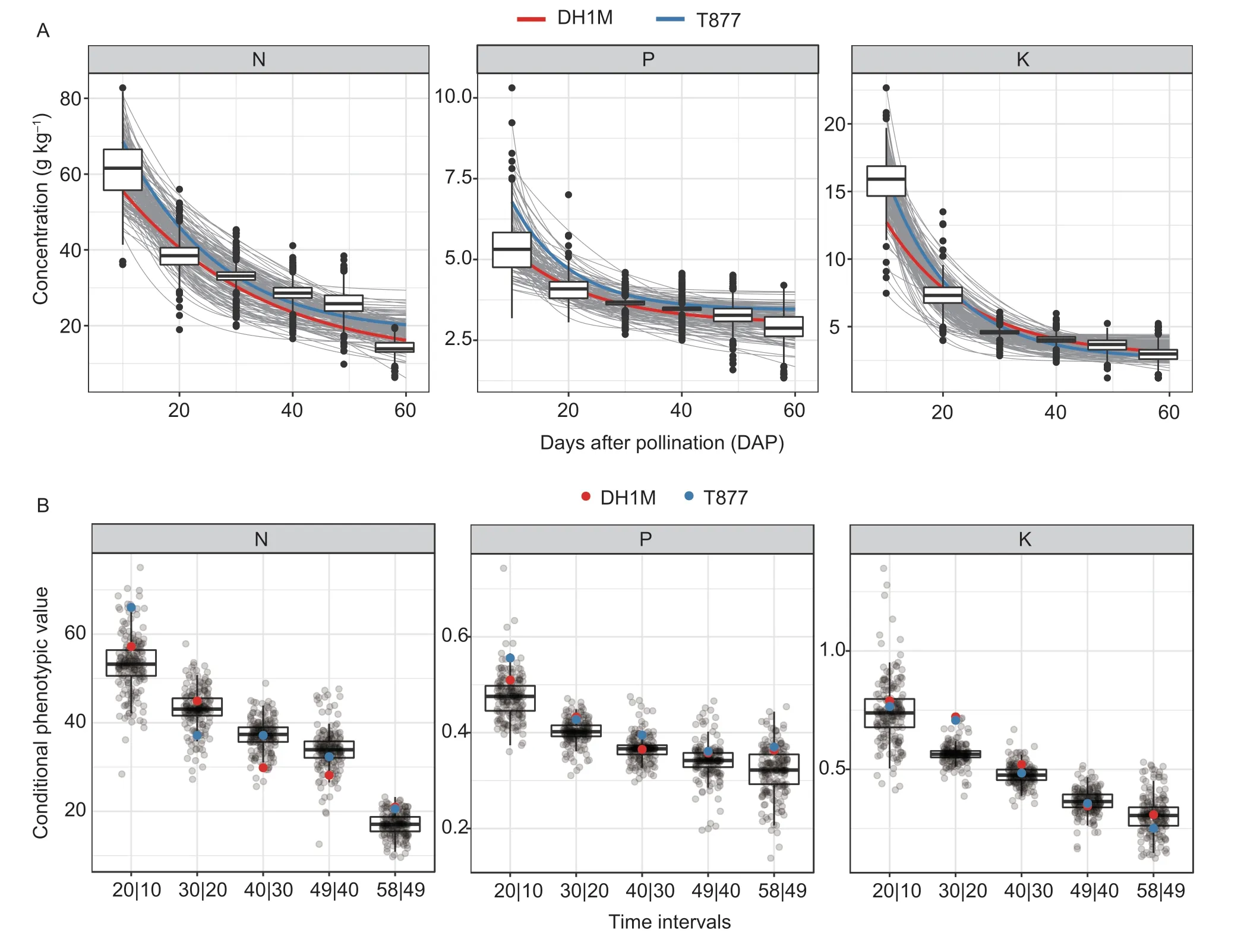
Fig. 1 The processes of changes in N,P and K contents and net changes during the seed developmental process in maize. A,trajectories of changes in the N,P and K contents during the seed developmental process. Red and blue lines represent trajectories of changes in the N,P and K contents of DH1M and T877,respectively. B,distribution of conditional phenotypic values for N,P and K at different time intervals. Red and blue points represent the net changes of DH1M and T877,respectively. X-axis indicates the time intervals represented as t|t–1,and t is the number of days after pollination.
3.2.Unconditional QTLs associated with N,P,and K accumulation
QTL mapping based on the N,P,and K concentrations at the six time points identified 41 non-redundant QTLs,including 17 associated with N,16 associated with P,and 14 associated with K (Appendices E and F). As shown in Fig.2-A,each nutrient exhibited obviously different peaks at different time points on the LOD curve. Some QTLs were repeatedly detected at different time points,such as a QTL for N(10) and N(49) at 55.84 cM on chromosome 1,a QTL for P(40) and P(49)at 106.59 cM on chromosome 4,and one for K(40)and K(49) at 182.15 cM on chromosome 6. We also detected several pleiotropic QTLs for different nutrients,such as a QTL located at 14.36 cM on chromosome 4 controlling both P(49) and N(49),and a QTL located at 147.81 cM on chromosome 5 controlling both P(20)and N(20). Some pleiotropic QTLs were also detected at different time points. For example,a QTL located at 106.83 cM on chromosome 7 controlling both P(58)and K(58) was also associated with the concentration of K(30). The phenotypic variation explained (PVE) by each QTL across the six time points ranged from 2.01 to 7.95% for N,1.72 to 9.48% for P,and 1.21 to 13.18%for K (Fig.2-B;Appendix E). Among the identified QTLs,a similar proportion of QTLs carried the favorable allele originating from either one of the parental lines,T877 or DH1M. Nevertheless,T877 was the source of all increasing alleles contributing to N(49),whereas all QTL alleles that increased N(58),P(40),K(30) and K(58)originated from DH1M (Fig.2-C).
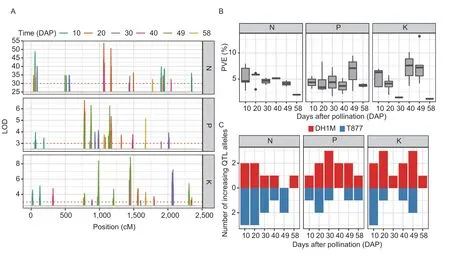
Fig. 2 Unconditonal QTL mapping of the N,P and K contents at six time points. A,LOD curves of the N,P and K contents at different time points. B,phenotypic variation explained (PVE) distribution of corresponding QTLs for the N,P and K contents at different time points. C,number of increasing QTL alleles for the N,P and K contents contributed by each parent. Blue and red represent the number of increasing QTL alleles contributed by T877 and DH1M,respectively.
3.3.Conditional QTLs associated with net changes in N,P,and K
Conditional QTL mapping identified 39 non-redundant QTLs contributing to the net effects of nutrient concentrations between adjacent time points (Fig.3-A;Appendices E and F). Regarding the net effects of the N concentration at different time intervals,we detected 12 associated QTLs,with PVEs ranging from 0.93 to 10.34% (Fig.3-B). One to five QTLs were detected for the net effects of N concentration (Fig.3-C). In addition,we detected 11 QTLs associated with the net effects of P concentration,with PVEs of 1.85 to 8.14% (Fig.3-B). In the first time interval (from 10 to 20 DAP),T877 was the source of all QTL alleles responsible for increasing the P content,whereas DH1M provided all increasing QTL alleles in the time interval from 30 to 40 DAP. Moreover,we detected 17 QTLs associated with the net effects of K concentration,with PVEs ranging from 1.03 to 10.32% (Fig.3-B). Two of these loci,on chromosome 6,had significant effects at multiple time intervals: a locus located at 182.15 cM controlling both K(49|40) and K(58|49),and a locus located at 293.03 cM controlling both K(20|10) and K(49|40). During the final time interval(from 49 to 58 DAP),T877 provided all QTL alleles for the increased K concentrations (Fig.3-C). Accounting for both unconditional and conditional QTLs,a total of 18 colocated QTLs,associated both with nutrient accumulation and net changes in concentration,were found throughout the seed developmental process (Appendix G). It was a locus at 182.15 cM on chromosome 5 controlling K(40),K(49),K(49|40),and K(58|49);a locus at 121.15 cM on chromosome 4 controlling K(10),K(40),and K(40|30);a locus at 219.92 cM on chromosome 4 controlling K(10),K(49),K(49|40),and P(30);a locus at 147.81 cM on chromosome 5 controlling N(20),N(20|10),and P(20);and a locus at 106.83 cM on chromosome 7 controlling K(30),K(58),P(58),and P(58|49).
Based on these unconditional and conditional loci associated with the N,P,and K concentrations,the RILs could be clearly divided into five clusters (Fig.4). We then analyzed the differences in nutrient concentrations among the different clusters during seed development.Significant differences in the N concentration were observed among the clusters at all time points except for 40 DAP. Members of cluster 4 had high N concentrations at all time points except for 10 DAP. The concentrations of P were significantly different among the clusters at all time points other than 10 DAP. The RILs grouped in cluster 4 had high P concentrations at all time points except for 10 DAP. No significant differences in K concentration were evident among the five clusters at 10 or 58 DAP. Cluster 2 had the highest K concentration at all time points except for 10 DAP.

Fig. 3 Conditonal QTL mapping of N,P and K net changes at different time intervals. A,LOD curves of N,P and K net changes at different time intervals. B,phenotypic variation explained (PVE) distribution of the corresponding QTLs for N,P and K net changes at different time intervals. C,number of increasing QTL alleles for the N,P and K net changes contributed by each parent. Blue and red represent the number of increasing QTL alleles contributed by T877 and DH1M,respectively. X-axis indicats the time intervals represented as t|t–1,and t is the number of days after pollination.
3.4.Combined multi-omics analysis to identify candidate genes
To examine the dynamics of gene expression during grain development,we compared T877 and DH1M at 10,20,30,40,and 50 DAP (Fig.5-A;Appendix H). The number of DEGs ranged from 1,465 (10 DAP) to 6,980(40 DAP). Because the transport of N,P,and K plays a vital role in nutrient uptake,transport,and remobilization,we were particularly interested in the expression of genes encoding nitrate transporter (NRT),nitrate reductase (NR),phosphate transporter (PHT),and potassium transporter(KT) in the different accessions. A total of 48 nutrientassociated DEGs were identified,including six for NRT,four for NR,16 for PHT and 22 for KT (Fig.5-C). Among the nutrient concentration-related QTLs,767,724,and 902 DEGs were associated with the concentrations of N,P,and K,respectively (Fig.5-D). To investigate the genes related to N,P,and K concentrations during grain development,we used WGCNA to identify gene modules at five developmental stages (Fig.5-B;Appendix I).A total of 37 modules were identified by WGCNA. A module-trait correlation analysis revealed 9,7 and 8 modules significantly associated with the N,P,and K concentrations,respectively (Fig.5-B). A gene–trait correlation analysis was also conducted for the genes in these modules,which uncovered 368,254,and 255 genes significantly correlated with the N,P,and K concentrations (Fig.5-D).
To determine the genomic differences between T877 and DH1M,we performed genomic re-sequencing of the two genotypes. We compared the genomes of T877 and DH1M to identify genome-wide single-nucleotide polymorphisms (SNPs). After filtering out low-quality SNPs,we obtained 489,668 SNPs between T877 and DH1M;of these,19,155 SNPs were in exons,including 8,598 SNPs representing nonsynonymous substitutions and 100 and 19 respectively leading to the gain or loss of a stop codon (Appendix J). These SNPs may have significant effects on gene function. A total of 44,36,and 44 DEGs with nonsynonymous substitutions between T877 and DH1M were associated with the concentrations of N,P,and K,respectively (Fig.5-D).

Fig. 4 Classification of clusters related to the N,P and K contents in the recombinant inbred line (RIL) population. A,C and E show cluster heat maps based on QTLs associated with the N,P and K contents,respectively. Blue and red represent the number of increasing QTL alleles contributed by T877 and DH1M,respectively. B,D and F show box plots of the N,P and K contents at different time points,respectively. Significant difference between clusters is indicated by different letters.

Fig. 5 Identification of candidate genes by QTL mapping,gene expression,co-expression analysis and comparative genomics for the concentrations of N,P and K. A,number of differentially expressed genes between T877 and DH1M. B,heatmaps showing the correlations of module-trait. The Pearson correlation coefficients and P-values of significant modules are given. C,expression levels of nitrate transporter (NRT),nitrate reductase (NR),phosphate transporter (PHT) and potassium transporter (KT) in different accessions. D,QTL candidate gene prediction process. E,distributions of QTLs (a,c and f for N,P and K) and candidate genes(b,d and f for N,P and K). The bars indicate the correlation coefficients between the gene expression level and the N,P and K concentrations,red and green bars indicate positive and negative correlations,respectively.
Using a combination of QTL,gene expression,coexpression analysis,and comparative genomic data,we identified 44,36,and 44 candidate genes related to N,P,and K accumulation,respectively (Appendices K,L and M).GRMZM2G371058,the candidate gene for qN20-6and qN20|10-6associated with the N concentration at 20 DAP and N(20|10) (Fig.6-A;Appendix K),was predicted to encode a Dof-type zinc finger DNA-binding family protein,and its expression level was significantly different between T877 and DH1M at 30 and 50 DAP.GRMZM2G371058,in the WGCNA turquoise module,was significantly correlated with the N concentration (r=0.92;P=1.5×10–6;Fig.6-C;Appendix K). Two nonsynonymous SNPs were observed in the first exon ofGRMZM2G371058(Fig.6-B). The candidate gene for qN40|30-1was associated with N(40|30),andGRMZM2G118646encoding a degradation of periplasmic protein,was significantly differentially expressed between T877 and DH1M at 40 DAP (Appendix K).GRMZM2G118646was also in the turquoise module and was significantly correlated with the N concentration (r=0.61;P=0.015).One nonsynonymous SNP was present in exon 2 of this gene. qP30-4-1was associated with the P concentration at 30 DAP. Its candidate gene,GRMZM2G422750encoding a chalcone and stilbene synthase family protein,was significantly differentially expressed between T877 and DH1M at four time points (all except 10 DAP).GRMZM2G422750,in the WGCNA black module,was significantly correlated with the P concentration (r=0.83;P=1.29×10–4),and one nonsynonymous SNP was found in exon 2 of this gene. Among the candidate genes,two genes (GRMZM2G159399andGRMZM2G317900)encoding an auxin response factor and one gene(GRMZM2G074427) encoding an auxin/indole-3-acetic acid (Aux/IAA) transcription factor,were associated with the P concentration (Appendix L). qK20|10-4was associated with K(20|10) (Fig.6-D). The candidate gene for qK20|10-4wasGRMZM2G113967encoding a CBL-interacting protein kinase,and the correlation coefficient between the expression level of this gene and the K concentration was 0.52 (Fig.6-F). Two nonsynonymous SNPs were observed in the first exon ofGRMZM2G113967(Fig.6-E).The candidate genes for K concentration includedGRMZM2G305526andGRMZM5G814718encoding laccase,GRMZM2G370991encoding an NRAMP metal ion transporter family protein,andGRMZM2G156599encoding the protein YELLOW STRIPE like 3 (Appendix M).

Fig. 6 QTLs and candidate genes for N and K on chromosomes 6 and 4,respectively. A and D,regional QTL plot surrounding the peak signals on chromosome 6 for N_20 and N(20|10) (A) and chromosome 4 for K(20|10) (D). B and E,gene structures and functional variations of GRMZM2G371058 (B) and GRMZM2G113967 (E). C and F,correlation analyses between gene expression levels of GRMZM2G371058 (C) and GRMZM2G113967 (F) and concentration of N (C) and K (F).
4.Discussion
Enhancing the grain accumulation of N,P,and K,three essential macronutrients in maize,is not only important for plant growth and development but it is also crucial for crop yield and quality. The detection of favorable QTLs/genes is a key step for improving nutrient concentrations by molecular breeding (Guet al.2015;Huanget al.2022).The accumulation of nutrients in kernels is a dynamic process. Therefore,we first measured the N,P,and K concentrations at six time points during seed development and determined the net changes in these nutrients at each time interval. Next,we detected unconditional and conditional QTLs associated with the N,P,and K concentrations. Finally,we integrated QTL mapping,gene expression,co-expression analysis,and genetic variance data,and identified 44,36,and 44 candidate genes,respectively,underlying QTLs for the N,P and K contents during maize grain development.
4.1.Phenotypic variation of the N,P,and K concentrations during seed development in maize
In this study,the concentrations of all three nutrients gradually decreased from 10 to 58 DAP. Other studies on changes in nutrient concentrations in developing grains have yielded divergent results (Alfoldiet al.1994). For example,Alfoldiet al.(1994) also reported that nutrient concentrations generally declined over time(Alfoldiet al.1994);however,they found thatopaque-2endosperm hybrids exhibited a slight increase in grain N concentration,which was the opposite of that observed in normal maize hybrids (Hirelet al.2007). In another study,the P concentration of tropical germplasm increased strongly during grain filling (Feilet al.1993). These inconsistent results may be due to the use of different genotypes. Considerable phenotypic variation exists in maize regarding nutrient concentrations. For example,the N concentration of teosinte lines is approximately 30%,whereas that of maize inbred lines ranges from 6.5 to 16.5% (Huanget al.2022). In the present study,the N,P,and K concentrations at 58 DAP varied from 6.32 to 19.56%,1.3 to 4.2%,and 1.2 to 5.23%,respectively.Nutrients accumulating in maize grains are derived from post-silking uptake and remobilization from other vegetative organs during the pre-silking stage (Hirelet al.2007;Chenet al.2016). Mineral nutrients can be roughly classified by mobility into three categories: highly mobile(e.g.,N,P,K,and Mg),moderately mobile (e.g.,Fe,Cu,Zn,and B),and weakly mobile (Ca and Mn) (Clarkson and Hanson 1980;Chenet al.2016). Among highly mobile nutrient elements,N and P are remobilized from all vegetative organs (44–51% for N and 38–60% for P) (Chenet al.2016),whereas K is mainly remobilized from leaves (51%). Unfortunately,we had no data on the concentrations of N,P,and K in leaves and stalks in this study that could have been used to investigate nutrient remobilization from different tissues to grain at the genetic level. Among the DEGs between parental lines,however,we identified 48 DEGs,including six NRTs,four NRs,16 PHTs,and 22 KTs,that suggest possible differences in post-silking nutrient uptake and remobilization between T877 and DH1M. The concentration of nutrients in mature grains is also determined by the dilution from carbohydrate deposition (Alfoldiet al.1994). To assess the influence of this dilution effect on grain nutrient concentrations,we compared the QTLs identified in this study with QTLs for seed-filling characteristics uncovered in the same RIL population (Yinet al.2020). A total of 14 QTLs (~22.2%) from the present study overlapped with QTLs for seed dry weight,filling rate,and seed-filling duration,which indicates that nutrient accumulation in grains is not completely consistent with the accumulation of dry matter in this study (Alfoldiet al.1994).
4.2.Unconditional and conditional QTLs for the N,P,and K concentrations
Unconditional and conditional QTL mapping respectively uncovered 41 and 39 QTLs for the N,P,and K concentrations. Most QTLs could explain less than 10%of the phenotypic variation,which indicates that these traits are controlled by many small-effect QTLs in our population. A total of 14 QTLs were detected by both mapping strategies. The discovery of these co-located QTLs based on both unconditional and conditional mapping indicate that the concentrations and net changes are not independent and are affected by the same loci,which is similar to the developmental features of other traits,such as tiller number (Liuet al.2010),plant height(Wanget al.2015),and seed hardness (Buet al.2018).In addition,eight QTLs were repeatedly detected at various time points,which is interesting because the temporal and spatial specificity of gene expression limits the detection of most QTLs to only one stage (Zhenget al.2011). Only three QTLs were associated with two nutrients,namely,two for P and K and one for P and N,which indicates that the accumulation of N,P,and K during maize grain development is regulated by different QTLs/genes (Renet al.2023).
4.3.Candidate genes for the concentrations of N,P,and K
Multi-omics analysis has played an important role in the mining of genes related to high-efficiency nutrient utilization in crops (Gonget al.2020;Liet al.2020;Duet al.2021). In the present study,44,36,and 44 candidate genes for the concentrations of N,P,and K,respectively,were identified by combined QTL,gene expression,co-expression,and comparative genomic analyses. One of these genes,GRMZM2G371058encoding a Dof-type zinc finger DNA-binding family protein,was associated with the N concentration at 10 DAP and N(20|10).Dofgenes are involved in N metabolism in several crops and can improve protein content. In rice,OsDOF18mediates ammonium transport and N distribution,in turn affecting N use efficiency (Wuet al.2017). In wheat and millet,the gene expression profiles ofTaDof1andEcDof1coincide with the enzymatic activities of GS and GOGAT,andEcDof1controls grain protein content (Kumaret al.2009;Guptaet al.2013).Another gene,GRMZM2G118646encoding a degradation of periplasmic protein,was associated with N(40|30). The homologous gene inArabidopsisthaliana,DEG5,forms a hexamer withDEG8,which is involved in the cleavage of photodamaged D2 protein of photosystem II (Sunet al.2007). The expression level ofDEG5,which is downregulated during leaf senescence,may play a role in N remobilization (Havéet al.2017). In the present study,we identified several genes related to P concentration that are involved in the auxin signaling pathway,including two auxin response factors (ARFs) and one Aux/IAA transcription factor. Previous studies have found thatZmARF4andZmARF31confer P tolerance by promoting root development (Wuet al.2016;Liet al.2022).GRMZM2G113967encoding a CBL-interacting protein kinase was associated with the K concentration in our study. Many K+channels and transporters are regulated by CBL calcium binding proteins and their interacting protein kinases,the CIPKs (Zhanget al.2023). In maize,ZmCIPK23 interacts with ZMK1 and phosphorylates the cytosolic region of ZMK1,thereby activating ZMK1-mediated K+uptake. ZmCBL1-ZmCIPK23 can also activate ZmHAK5-mediated K+uptake (Zhanget al.2023).In cassava,MeCIPK10regulates the transition of the K+transport activity ofMeAKT2(Chenet al.2023). These candidate genes may play a role in N remobilization,but their functions need to be investigated using mutants and overexpressing lines.
5.Conclusion
We evaluated the grain N,P,and K concentrations in 206 recombinant inbred lines generated from a cross of DH1M and T877 at six time points after pollination. A total of 41 and 39 unconditional and conditional QTLs for the N,P,and K concentrations were detected by QTL mapping. Through a combined multi-omics analysis,we identified 44,36,and 44 candidate genes for the N,P,and K concentrations,respectively. These results provide the most promising loci and genes for improving nutrient concentrations in maize.
Acknowlegements
This work was supported by the Seed Industry Revitalization Project of Jiangsu Province,China(JBGS[2021]009),the National Natural Science Foundation of China (32061143030 and 31972487),the Jiangsu Province University Basic Science Research Project,China (21KJA210002),the Key Research and Development Program of Jiangsu Province,China (BE2022343),the Innovative Research Team of Universities in Jiangsu Province,China,the High-end Talent Project of Yangzhou University,China,the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD),China,and the Qing Lan Project of Jiangsu Province,China.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2023.11.003
杂志排行

Journal of Integrative Agriculture的其它文章
- Molecular mechanisms of stress resistance in sorghum: lmplications for crop improvement strategies
- Artificial selection of the Green Revolution gene Semidwarf 1 is implicated in upland rice breeding
- The NAC transcription factor LuNAC61 negatively regulates fiber development in flax (Linum usitatissimum L.)
- The underlying mechanism of variety–water–nitrogen–stubble damage interactions on yield formation in ratoon rice with low stubble height under mechanized harvesting
- Rice canopy temperature is affected by nitrogen fertilizer
- The first factor affecting dryland winter wheat grain yield under various mulching measures: Spike number