硼砂煅制工艺优化及质量评价
2024-03-12杨辛欣洪禹昕赵晶丽王楚盈许天阳
杨辛欣,洪禹昕,张 睿,赵晶丽,刘 佳,王楚盈,许天阳,于 澎
(长春中医药大学,吉林 长春 130117)
硼砂始载于五代《日华子本草》[1],是由天然矿物硼砂经精制而成的结晶体,具有清热解毒等功效,可用于治疗咽喉肿痛、口舌生疮等,主要成分为含水四硼酸钠,其炮制方法众多,例如宋代记载“研”[2]、“熬”[3]; 元代使用“油浸法”[4]; 明代大多沿用前人方法,并且后期出现 “另研”[5]、“研如飞尘”[6]; 清代典籍提到“煅”[7]等。煅制可除去结晶水,减小用药刺激,增强燥湿收敛、促进溃疡愈合的作用,所谓 “生则化腐,煅枯则生肌”[8],但历版《中国药典》 均未收录硼砂标准,全国各地药材标准及炮制规范中其煅制工艺规定也较简单,相关参数及炮制终点并无明确规定,导致炮制品极易出现“结底” “硬心” “污边” 等问题,从而大批量生产成品率低、质量差异大。
本实验在四硼酸钠含量、失水率[9]的基础上,根据炮制目的增加蓬松度、粉碎率2 个指标,采用星点设计-效应面法对硼砂煅制工艺进行优化,同时采用扫描电镜、热重分析、拉曼光谱、X 射线衍射进行质量评价,该方法可解决该药材炮制工艺混乱、终点不明确等问题,为判断其质量提供依据。
1 材料
1.1 仪器 MC02810218 型马弗炉(余姚市金电仪表有限公司); FA1204B 型电子天平(万分之一,上海精密科学仪器有限公司); SU8020 型场发射扫描电子显微镜镜(日本日立公司); STA 449 F5 Jupiter 型热重分析仪 (德国Netzsch 公司);inVia 型拉曼光谱仪 (英国Renishaw 公司); D8 Advance X 型射线粉末衍射仪(德国Bruker 公司);JG500 g 型粉碎机(上海广沙工贸有限公司); 50 mL 聚四氟乙烯滴定管(天津市天玻玻璃仪器有限公司); VTO-34A 型热风电烤箱[北美电器(珠海) 有限公司]; 耐热玻璃(山东业盛玻璃有限公司)。
1.2 试剂 盐酸(分析纯,批号20180828)、氢氧化钠(分析纯,批号20170511) (北京化工厂有限责任公司); 甲基红(分析纯,批号20180927)、甲基橙(分析纯,批号20181123) (天津市光复精细化工研究所); 酚酞(分析纯,批号20190224,上海化学试剂分装厂)。
1.3 药材 硼砂(批号20161201,产地河南) 购于河北顺全隆药业有限公司,经长春中医药大学中药鉴定教研室翁丽丽教授鉴定为正品。
2 方法与结果
2.1 评价指标测定
2.1.1 失水率 采用质量法,煅制前计为M1,煅制后计为M2,公式为失水率=[(M1-M2) /M1] ×100%。
2.1.2 蓬松度 称取一定质量(M) 煅硼砂(过5 号筛) 装在量筒中,反复振动至其体积不再下降,记录体积(V),公式为蓬松度=V/M。
2.1.3 粉碎率 称取一定质量(M1) 煅硼砂,将其放入粉碎机粉碎20 s,冷却至室温后过6 号筛,收集粉末,称定质量M2,公式[10]为粉碎率=(M2/M1) ×100%。
2.1.4 四硼酸钠含量测定 取煅硼砂约0.4 g,精密称定,置于50 mL 锥形瓶中,加25 mL 水溶解,再加0.05%甲基红指示剂1 滴,盐酸滴定液(0.1 mol/L) 滴至溶液变为橙红色 (每1 mL 相当于10.06 mg 四硼酸钠)。
2.2 煅制工艺优化 在预实验基础上,选择硼砂铺设厚度 (X1)、煅制时间 (X2)、煅制温度(X3) 作为影响因素,根据星点设计原理,每个因素分别设置5 个水平,用代码值-α、-1、0、1、α表示(α =1.732),具体见表1。

表1 星点设计-效应面法因素水平Tab.1 Factors and levels for central composite designresponse surface method
采用马弗炉对硼砂进行煅制,通过感温元件及温控器对马弗炉进行温度调控和检测,按表1 因素水平开展实验,测定失水率(Y1)、蓬松度(Y2)、粉碎率(Y3)、四硼酸钠含量(Y4),结果见表2。

表2 星点设计-效应面法设计及结果Tab.2 Design and results for central composite design-response surface method
采用Design Expert 7.0 软件对表2 数据分别进行多元线性回归、二项式拟合,得线性方程分别为Y1=39.55-0.91X1+2.61X2+7X3(R2=0.735 3,P<0.05)、Y2=0.476 42-0.037 831X1+0.158 24X2+0.001 74X3(R2=0.733 0,P<0.05)、Y3=89.95-0.56X1+4.04X2+5.66X3(R2=0.312 0,P<0.05)、Y4=58.292 91-2.126 32X1+7.109 25X2+0.083 91X3(R2=0.724 5,P<0.05); 二项式方程分别为Y1=26.979 5X2+0.307X3-8.487 22X22-0.000 453X32(R2=0.919 2,P<0.05)、Y2=0.042 297 +0.791 62X2+0.001 74X3- 0.255 39X22(R2=0.791 0,P<0.05)、Y3=5.725 41 +85.590 78X2+0.181 96X3-30.808 08X22-0.000 234X32(R2=0.579 5,P<0.05)、Y4=1.485 26-2.126 32X1+39.859 4X2+0.408 76X3- 0.044 174X2X3-8.761 62X22- 0.000 541X32(R2=0.975 2,P<0.05)。由此可知,各评价指标P值均小于0.05,即对模型均有显著影响,但二项式方程相关系数R2显著高于线性方程,表明前者拟合度更高,预测性更好,分析方法更可靠。
根据上述二项式方程考察各因素对煅制工艺的影响,结合响应面图(图1 ~4),选择失水率、蓬松度、粉碎率、四硼酸钠含量最大的区域,最佳取值范围见表3。

图1 煅制温度、煅制时间对硼砂失水率的影响Fig.1 Effects of calcining temperature and calcining time on the water loss of Borax

图2 煅制温度、煅制时间对硼砂蓬松度的影响Fig.2 Effects of calcining temperature and calcining time on the fluffiness of Borax
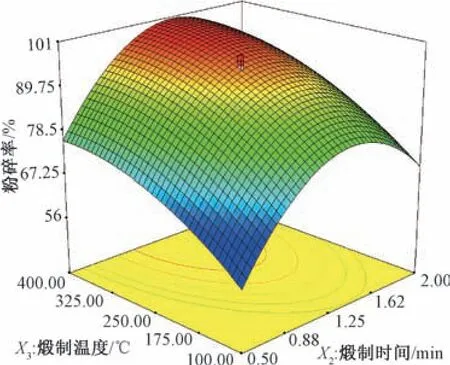
图3 煅制温度、煅制时间对硼砂粉碎率的影响Fig.3 Effects of calcining temperature and calcining time on the crushing rate of Borax
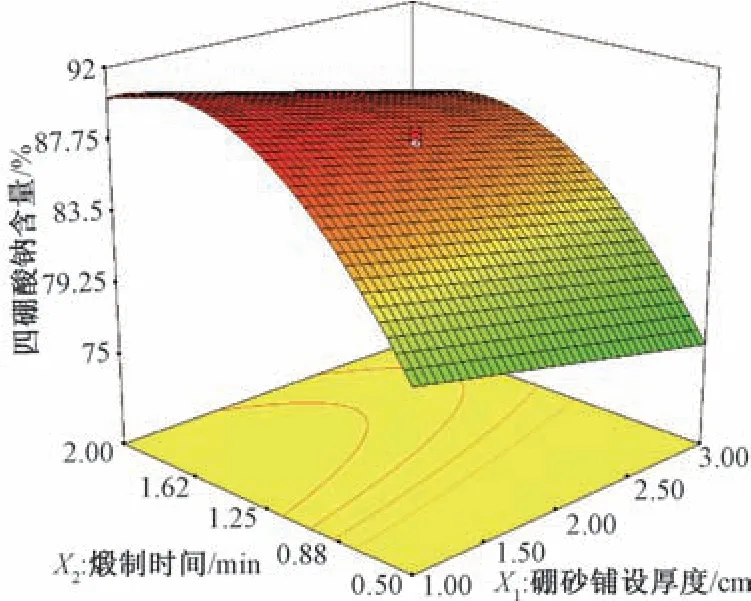
图4 硼砂铺设厚度、煅制时间对四硼酸钠含量的影响Fig.4 Effects of Borax laying thickness and calcining time on sodium tetraborate content
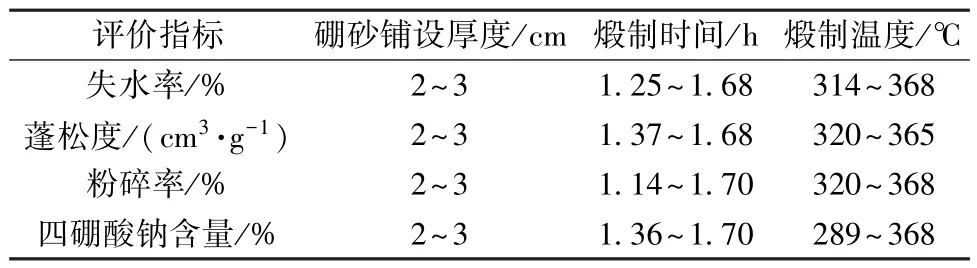
表3 各影响因素最佳取值范围Tab.3 Optimal value ranges of various influencing factors
最终确定,最优煅制工艺为硼砂铺设厚度2 cm,煅制时间1.67 h,煅制温度365 ℃,考虑到工艺参数可控性,将其修正为取净硼砂适量,置于适宜容器内铺设均匀,铺设厚度约2 cm,武火(约365 ℃) 加热,煅至无水气挥发,呈白色酥松块状,时间约为100 min,取出放冷。根据上述工艺进行3 批验证试验,结果见表4,可知实测值与预测值的相对误差较小,重复性良好,表明该模型预测性良好,工艺稳定可行。
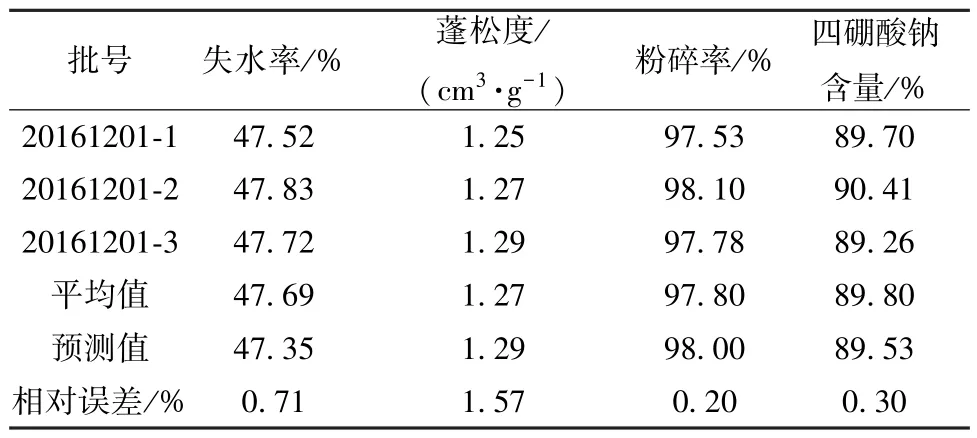
表4 验证试验结果(n=3)Tab.4 Results for verification tests (n=3)
2.3 质量评价 采用扫描电镜、热分析法、拉曼光谱、X 射线衍射技术,从微观性状、成分结构变化等方面进行考察,同时取原药材、按“2.2.3”项下最优工艺(煅制厚度、煅制温度不变,煅制时间30 min) 制备的中间品、按“2.2.3” 项下最优工艺制备的煅制品 (批号20161201-1) 适量,分析其动态变化。
2.3.1 微观形态 设置加速电压为3.0 kV,样品工作距离为7 mm 左右,放大倍数50、600、2 000、5 000 倍。蘸取少量原药材、中间品、煅制品粉末,均匀洒在金属柱面的导电胶带上,吹去多余粉末,调整扫描电子显微镜参数,收集图像,结果见图5~7。

图5 原药材扫描电镜图Fig.5 Scanning electron microscope image of original medicinal material
由图5 可知,原药材呈不规则块状结构,表面较平整光滑,结构致密,可见横纵交错裂痕,偶见不规则凹陷。由图6 可知,中间品呈不规则形状碎块,表面粗糙、疏松、不平整特征明显,致密结构断裂成大小不一的碎片,大碎片呈块状立体结构,小碎片呈片状立体结构。由图7 可知,煅制品呈圆形颗粒及不规则形状碎块,与中间品比较碎块体积更小,表面疏松多孔。

图6 中间品扫描电镜图Fig.6 Scanning electron microscope image of intermediate good

图7 煅制品扫描电镜图Fig.7 Scanning electron microscope image of calcined product
综上所述,硼砂煅制后微观形态发生变化,粒子体积减小,表面结构逐步疏松,可作为煅制终点判断的依据,并且煅制后疏松结构更有利于吸收细胞水分,减少炎症渗出物,从而增加消炎防腐的作用,在临床用药中更好地发挥其药效,方便外用及丸散剂制备。
2.3.2 热分析技术 设置升温速度为10 ℃/min,升温范围为0~1 100 ℃,气氛为静态空气,将原药材、中间品、煅制品粉碎后过5 号筛,程序升温,进行热谱扫描,各重复2 次,热重-微分热重(TGDTG) 曲线见图8。

图8 原药材(A)、中间品(B)、煅制品(C) TG-DTG 曲线Fig.8 TG-DTG curves for original medicinal material (A),intermediate good (B) and calcined product (C)
由图8A 可知,在53 ℃左右原药材开始失去结晶水而发生第1 次失重,随后在115 ℃左右开始发生第2 次失重,升温至212 ℃之后呈缓慢质量丢失现象,在750 ℃后曲线平缓直至失重结束,最终有64.17%的物质残留。由图8B 可知,中间品在141 ℃左右开始失去结晶水发生失重,升温至210 ℃后呈缓慢失重现象,在500 ℃后曲线平缓至失重结束,最终有79.71% 的物质残留。由图8C可知,煅制品未发生失重现象,说明采用最优工艺后在煅制过程中失去全部结晶水,从而起到了良好的提纯作用,有利于发挥其药效。
2.3.3 拉曼光谱 设置激发光源为785 nm,光谱测量范围为2 700 ~100 cm-1,激光功率为50 mV,光纤探头长度为1.5 m,激光强度为5%,扫描时间为3 000 ms。取原药材、煅制品粉末适量,置于样品槽中,压实,枪头轻抵两者表面进行光谱采集,平行3 次,采用Origin 8 软件绘制拉曼光谱图,结果见图9~10。
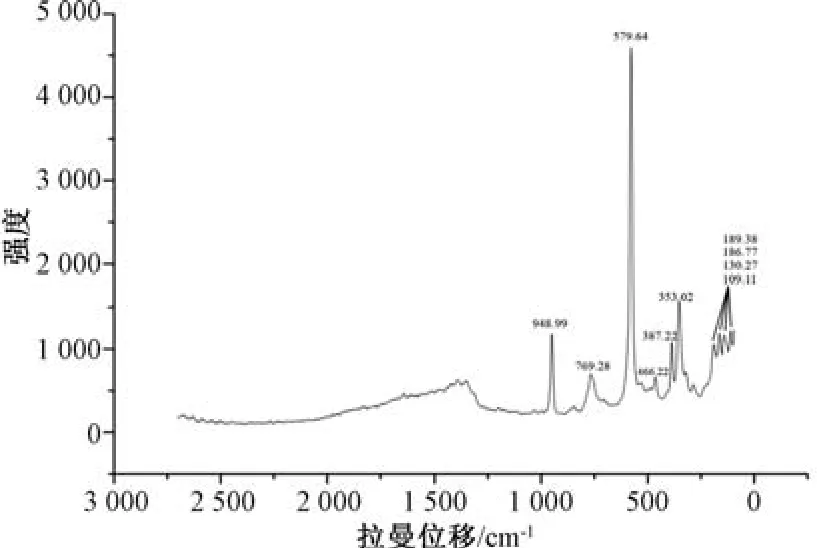
图9 原药材拉曼光谱图Fig.9 Raman spectrum for original medicinal material
前期报道,硼砂在拉曼光谱中的位移主要体现在四面体硼(BO45-)、三角形硼(B O33-)[11]、水分子、B (OH) 键[12]。由图9 可知,对于拉曼位移在579.64 cm-1处的峰,其峰值在4 818.27 的是四面体硼(BO45-) 振动最强特征峰,在466.22、387.22、353.02cm-1处,其峰值在714.82、1 211.69、1 635.05 的是四面体硼(BO45-) 对称弯曲振动中强特征峰; 在769.20 cm-1处,其峰值在726.39 的是四面体硼(BO45-) 对称伸缩振动;在948.99 cm-1处,其峰值在1 291.43的是三角形硼(BO33-) 对称伸缩振动; 在189.38、186.77、130.27、109.11 cm-1处,其峰值在1 095.74、1 039.99、1 153.09、1 293.04 的是晶格振动[13]。由图10 可知,在原药材相应拉曼位移处存在四面体硼(BO45-) 的特征峰,而且峰值有不同程度的变大,在三角形硼(BO33-) 处出现2 个峰,但在晶格振动的拉曼位移处仅产生1 个峰,表明煅制后硼砂原成分结构发生改变,其原因是在此过程中含水量降低,分子中氢键作用减弱,硼原子对称性增加,导致拉曼位移、吸收峰有差异,进一步显示硼砂经煅制后可在不改变药物主成分的基础上,起到去除部分结晶水而达到精制纯化的作用,最终提高其药效。

图10 煅制品拉曼光谱图Fig.10 Raman spectrum for calcined product
2.3.4 X 射线衍射分析 设置入射光源CuKα 辐射,Ni 片滤波,X 管工作电压40 kV,电流40 mA,连续扫描方式,扫描速度6°/min,扫描范围5°~90°。取原药材、煅制品适量,粉碎过筛,研匀后在X 射线衍射仪样品板上压平检测,将相关谱图导入JADE 6.0 软件,进行寻峰处理、物相分析及PDF 标准卡片匹配,采用Origin 8.0 软件绘图,结果见图11。
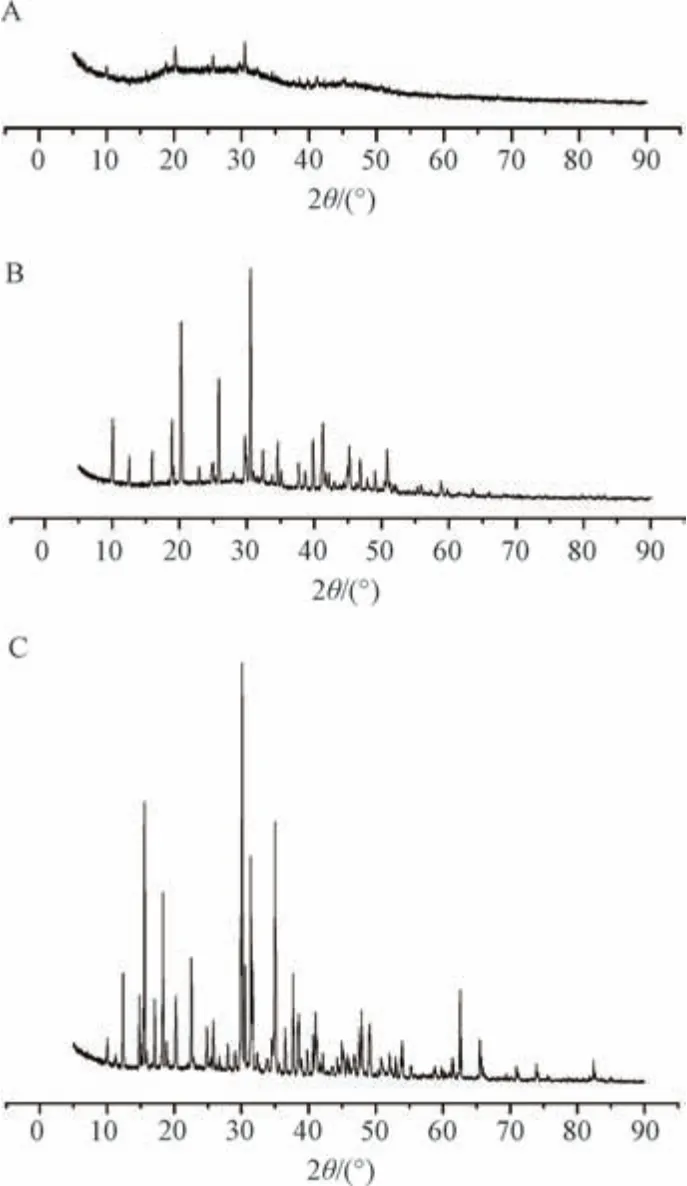
图11 原药材(A)、中间品(B)、煅制品(C) X 射线衍射图谱Fig.11 X-ray diffraction patterns of original medicinal material (A),intermediate good (B) and calcined product (C)
由图11A 可知,原药材主要成分为Na2B4O5(OH)4·8H2O,其特征强峰分别在2θ15.559°、18.329°、22.545°、30.089°、31.402°、31.607°、35.003°处,其对应晶面间距(相对峰强) 分别为0.569 (65.8)、0.484 (43.2)、0.394 (27.2)、0.297 (100.0)、0.285 (52.4)、0.283 (26.4)、0.256 (61.8),表明它为正品,而且纯度较高,与文献[14] 报道一致。由图11B 可知,中间品主要成分为Na2B4O7·5H2O,其特征强峰分别在2θ10.112°、18.892°、20.267°、25.853°、29.744°、30.559°、41.302°、50.840°处,其对应晶面间距(相对峰强) 分别为 0.874 (30.0)、0.469(30.0)、0.438 (76.6)、0.344 (49.2)、0.300(21.3)、0.292 (100.0)、0.218 (30.4)、0.179(19.8),表明它为五水硼砂,在365 ℃下煅制30 min 后其特征线逐渐消失,转化为五水硼砂特征线。由图11C 可知,煅制品在365 ℃下煅制100 min 后原药材特征线完全消失,表明其结构由晶态转变为非晶态,与文献[15] 报道一致。
综上所述,原药材在煅制过程中发生成分组成、晶型结构改变,原晶体Na2B4O5(OH)4·8H2O 逐渐失去结晶水而变为五水硼砂,最终失去所有结晶水,由晶体状态转变为非晶体状态,其原因可能是存在三价或四价硼原子振动,在无定形硼酸钠结构中包含稳定的-OH 基团,并与钠阳离子强键结合。结合电镜扫描、热分析结果,可知该工艺能有效提高硼砂主要成分四硼酸钠纯度,从而达到增加药效的作用,并对改善其用药稳定性、安全性具有积极意义。
3 讨论
3.1 煅制工艺优化 本实验以硼砂铺设厚度、煅制温度、煅制时间为影响因素,失水率、蓬松度、粉碎率、四硼酸钠含量为评价指标,采用星点设计-效应面法优化硼砂煅制工艺。验证试验结果表明,该工艺简便稳定,重复性好,精密度高,能综合考虑到各种因素之间的相互作用,并且所得产品质量可控,从而为中药企业相关工业化生产提供了参考。
3.2 质量评价 本实验采用扫描电镜,从微观角度上直观考察硼砂煅制前后的变化,发现煅制后其结构较疏松,更有利于在临床上发挥药效,也方便制备相关外用制剂和丸散剂,并且其微观形态的变化可作为煅制终点判断依据。然后,采用热分析法、拉曼光谱分析法、X 射线衍射法来观察硼砂煅制前后成分组成、晶体结构的变化,发现它在煅制过程中失去全部结晶水,有效成分四硼酸钠含量提高,从而起到提纯、除杂的作用。上述结果进一步确定硼砂最优煅制工艺的可行性与合理性,同时也能为其他矿物类中药的鉴别及质量评价提供思路。
