LiODFP在锂离子电池正极氧化分解的DFT研究
2024-03-11王雅婷林钰涵施志聪
王雅婷,林钰涵,刘 军,施志聪
(广东工业大学材料与能源学院新型电池研究所,广东 广州 510006)
锂离子电池的界面阻抗会随着循环次数的增加而变大,导致本体电解质的离子电导率下降,使得低温下电池的性能不理想[1]。作为锂离子电池电解液的常用锂盐,六氟磷酸锂(LiPF6)的热稳定性较差,在电池放充电过程中易发生分解产生LiF 和HF 等物质,导致界面阻抗增大,且腐蚀电极材料甚至集流体[2]。热稳定性好的锂盐能提高电池的热稳定性,甚至能在电极表面形成一层固态电解质相界面膜,提高电池电解液/电极界面的稳定性,成为锂离子电池的研究热点之一。如四氟盐酸锂(LiPF4C2O4)和二氟草酸硼酸锂(LiDFOB)可作为LiPF6的潜在替代品,以提高电池的热稳定性[3]。双(草酸)硼酸锂(LiBOB)和二氟二草酸磷酸锂(LiODFP)可在负极或正极上形成新的界面膜[4]。
LiODFP 是由LiPF6与草酸以及硅烷类物质合成的,因具有强的电化学稳定性和热稳定性而被关注。B.Liao 等[5]研究表明,制备石墨/LiNi0.5Co0.2Mn0.3O2电池时,在1 mol/L LiPF6/EC+EMC(质量比1∶2)电解液中加入LiODFP 添加剂,可以构建具有低阻抗的负极和正极界面薄膜,从而改善电池的低温性能。同样,LiODFP 添加剂可应用于高压锂离子电池(5 V LiNi0.5Mn1.5O4)中[6],形成正负极界面膜,防止金属离子从LiNi0.5Mn1.5O4正极中溶解析出后沉积在石墨表面。W.M.Zhao 等[7]用飞行时间质谱仪(TOF-MS)研究发现,LiODFP 添加剂在正极的成膜比负极成膜更厚。
LiODFP 对正极的保护作用比较明显。大多数的文献利用各种物理化学实验手段研究该添加剂在不同电池中的性能,但它的氧化分解机理仍不明确。本文作者通过密度泛函理论(DFT)来研究LiODFP 在正极表面的成膜机理。
1 计算方法
用Gaussian 09 软件中的DFT B3LYP/6-311++G(d)方法对氧化反应的反应物、中间体、过渡态及产物的结构进行优化[8]。通过振动频率,分析势能面上各驻点的性质,以确认优化得到的结构是势能面上的稳定点或是过渡态,以驻点虚频的唯一性确定反应过渡态。同时,使用内禀反应坐标(IRC)方法对过渡态进行跟踪计算,验证反应势能面上各过渡态与反应物和产物之间的连接关系。用自然键轨道(NBO)方法分析优化得到的分子中原子的电荷分布。考虑溶剂化效应对反应的影响,采用极化连续介质模型(PCM),在相同的计算水平上,研究体系在介电常数为20.5 的溶液中的反应,接近于实验中常用的线状碳酸酯与环状碳酸酯质量比为7∶3电解液的介电常数。氧化电势使用式(1)计算[9]:
式(1)中:G(M)和G(M+)分别是298.15 K 下,络合物M 以及其氧化态M+的吉布斯自由能值;F是法拉第常数;1.4 V 是指该氧化电势是相对于Li+/Li 的。
2 结果与讨论
2.1 锂盐阴离子单分子体系的氧化反应和电荷分布
一般而言,用于锂离子电池的无机锂盐普遍具有价格低、不易分解、能耐受高的电位、合成简单等优点。相对于无机锂盐,有机锂盐可认为是在无机锂盐的阴离子上又增加了吸电子基团调控而成。将锂盐LiODFP 与无机锂盐LiPF6、四氟硼酸锂(LiBF4)、高氯酸锂(LiClO4)和有机锂盐LiBOB、LiDFOB 作为对比,分析锂盐单分子氧化性能的规律。
根据分子轨道能量可知,最高占据轨道(HOMO)能量越大,分子越容易失去电子,从而越容易发生氧化;同样,最低未占据轨道(LUMO)能量越小,分子越容易得到电子,就越容易还原,能量间隙ELUMO-HOMO分子越小,越容易发生氧化还原反应。阴离子、碳酸酯分子及LiODFP 分子的HOMO 能量、LUMO 能量及ELUMO-HOMO见表1。

表1 不同锂盐阴离子与碳酸酯溶剂的前线轨道能量Table 1 Frontier orbital energy of different lithium salt anions and carbonate solvents
从表1 可知,除了ClO4-外,无机锂盐PF6-和BF4-的HOMO 能量都小于碳酸酯和有机锂盐阴离子(ODFP-、DFOB-、BOB-),即氧化性比碳酸酯和有机锂盐更稳定。这与文献中报道的实验结果一致,即锂盐的氧化稳定性为LiPF6>LiBF4>LiClO4[10-11]。LiClO4表现特殊,是因为Cl 处于最高价态+7,极易与电解液中的有机溶剂发生氧化反应,甚至引起电池燃烧和爆炸。ODFP-的HOMO 能量不仅远大于碳酸酯分子,也大于LiODFP。这表明在含LiODFP 碳酸酯电解液充电过程中,ODFP-阴离子比LiODFP 分子更容易氧化。
锂盐阴离子在氧化反应前后的优化结果和自然布居分析(NPA)电荷分布见图1,括号内为电荷变化的数量。
PF6-、BF4-、ClO4-在氧化前后,结构的变化主要体现在P-FB-FCl-O 的键长变化上;电荷的主要变化在F 和O 原子上,而P、B、Cl 的电荷变化微乎其微。ODFP-、DFOB-、BOB的氧化反应前后的结构变化比较明显,伴随着开环反应。

4.综合能源服务发展需要。成立区域检修公司,为开展综合能源服务提供可能性。在电力市场化改革情况下,可有效培育潜在电力用户。
已有文献报道,在锂盐存在的情况下,PC 的氧化电势降低[12]。L.D.Xing 等[13]使用DFT,研究锂离子电池电解液中多分子体系的氧化稳定性,发现锂盐阴离子和邻近溶剂分子的存在,会改变碳酸酯氧化反应的动力学及热力学过程,因此,阴离子对碳酸酯溶剂氧化稳定性的影响不容忽视。
2.2 碳酸酯-阴离子体系的氧化稳定性和电荷分布
碳酸酯(包括EC、PC、EMC 和DMC)的氧化电势见图2。
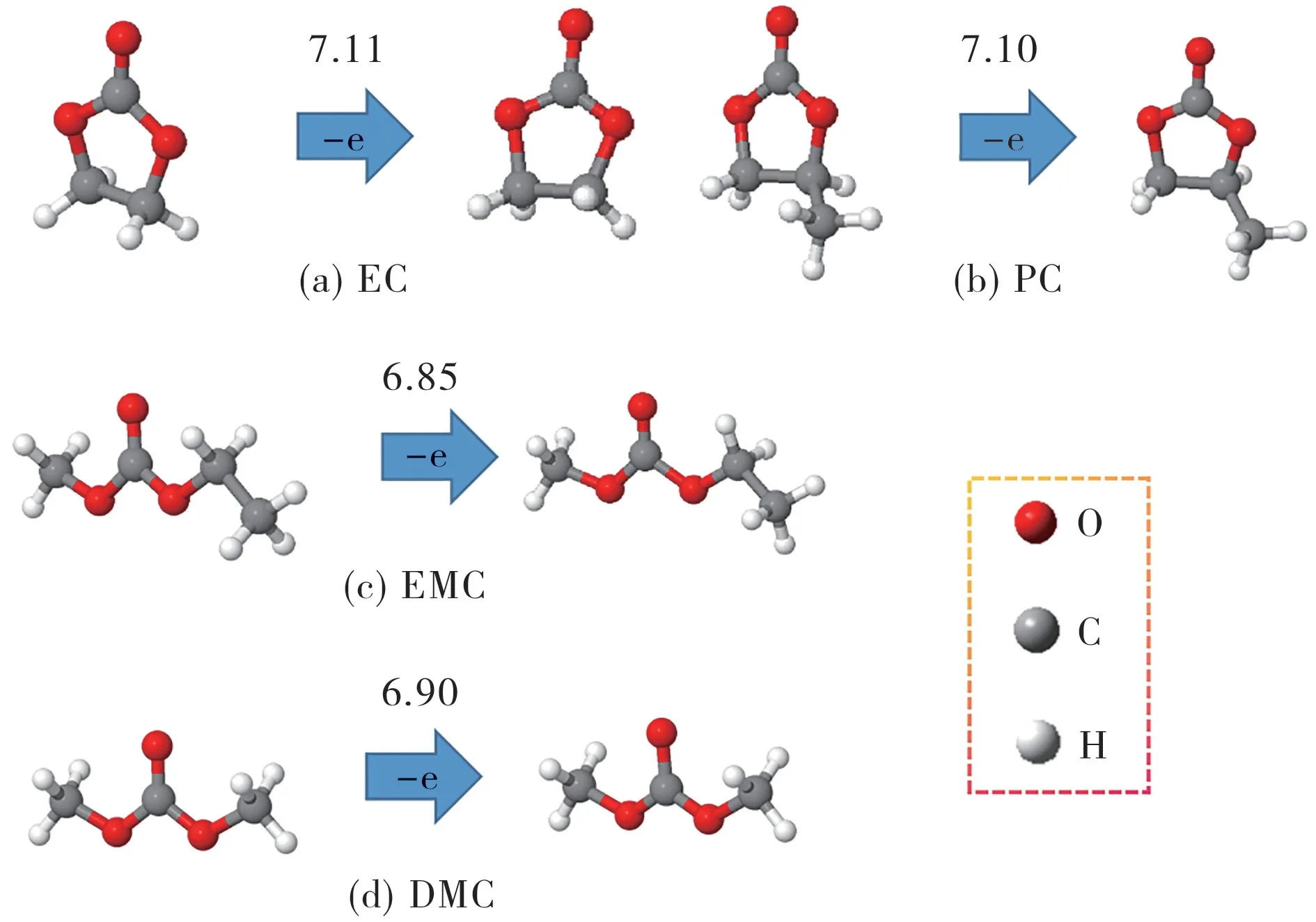
图2 碳酸酯的氧化电势(V,vs.Li/Li+)Fig.2 Oxidation potential(V,vs.Li/Li+)of carbonate
碳酸酯-ODFP-配合物、碳酸酯-DFOB-配合物、碳酸酯-BOB-配合物、碳酸酯-BF4-配合物、碳酸酯-ClO4-配合物、碳酸酯-PF6-配合物在氧化反应前后的优化结果和NPA 电荷分布见图3、图4。
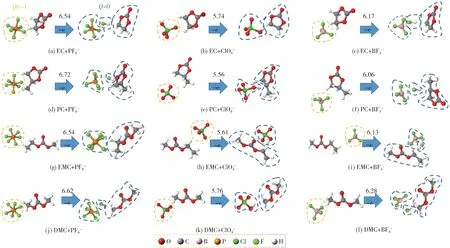
图3 EC/PC/EMC/DMC 与PF6-/BF4-/ClO4-的氧化电势(V,vs.Li/Li+)Fig.3 Oxidation potential(V,vs.Li/Li+)of EC/PC/EMC/DMC combining with PF6-/BF4-/ClO4-
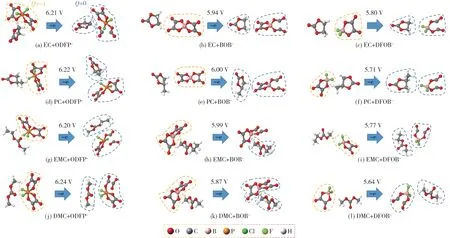
图4 EC/PC/EMC/DMC 与ODFP-/BOB-/DFOB-的氧化电势(V,vs.Li/Li+)Fig.4 Oxidation potential(V,vs.Li/Li+)of EC/PC/EMC/DMC combining with ODFP-/BOB-/DFOB-
从图2-4 可知,所研究碳酸酯和配合物的氧化电势排序:碳酸酯单分子>碳酸酯-PF6-配合物>碳酸酯-ODFP-配合物>碳酸酯-BF4-配合物>碳酸酯-BOB-配合物>碳酸酯-ODFB-配合物>碳酸酯-ClO4-配合物。只有DMC 例外,原因可能是它为高度对称的线性碳酸酯。可以看出,锂盐阴离子的存在,都会不同程度地降低碳酸酯的氧化电势。

2.3 EC+ODFP-配合物的氧化分解机理
单独的EC 与ODFP-失去一个电子后的分解过程如图5所示。

图5 EC 和ODFP-氧化分解反应的过渡态和产物的结构及能量(kJ/mol)Fig.5 Structure and energy(kJ/mol)of transition states and products of oxidative decomposition reaction of EC and ODFP-
EC+ODFP--e 络合物7 条可能存在的分解路径见图6。

图6 EC+ODFP-络合物氧化分解反应的过渡态和产物的结构及能量(kJ/mol)Fig.6 Structure and energy(kJ/mol)of transition states and products of oxidative decomposition reaction of EC+ODFP-complex
从图6 可知,路径Path1 与Path2 分别对应从ODFP-与EC 开始的分解反应。在EC+ODFP--e 络合物中,分解路径Path1 与ODFP--e 分解路径相似,且两个过渡态TS1 能量与中间产物M1 能量都很相近。继续分解ODFP-剩下的M1,经过路径Path3,生成M3 含两个CO2、R1 自由基与EC 分子,其过程的能垒以及产物的能量与单个ODFP--e 中路径Path2很相近。综合路径Path1 与Path3 的结果看,EC+ODFP--e 络合物中分解ODFP-路径与单个ODFP--e 很相似,说明EC 溶剂分子的存在几乎不会影响ODFP-的分解能量。另外,继续分解M1 中EC 分子,即EC 发生开环反应,分别经过路径Path4 与Path5,生成相同产物,相应的激发能分别为220.26 kJ/mol(TS4-M1)与219.41 kJ/mol(TS5-M1),均高于路径Path3 的激发态60.81 kJ/mol(TS3-M1)。这表明,进一步分解M1 中EC 比分解ODFP-更难。同时,尽管Path3 的激发态相对比较低,但也高于Path1 的反应能垒。
2.4 自由基终止反应
EC+ODFP--e 络合物的初始反应产物有CO2气体生成,与研究EC 基电解液氧化反应的实验结果吻合[11]。对EC+ODFP--e 络合物的初始反应产物R0 和R1 的终止反应进行研究。
EC+R1 络合物分解反应的过渡态和产物的结构及能量见图7;R1 所有可能发生的聚合反应见图8。

图7 EC+R1 络合物分解反应的过渡态和产物的结构及能量(kJ/mol)Fig.7 Structure and energy(kJ/mol)of transition states and products of dicomposition reaction of EC+R1 complex
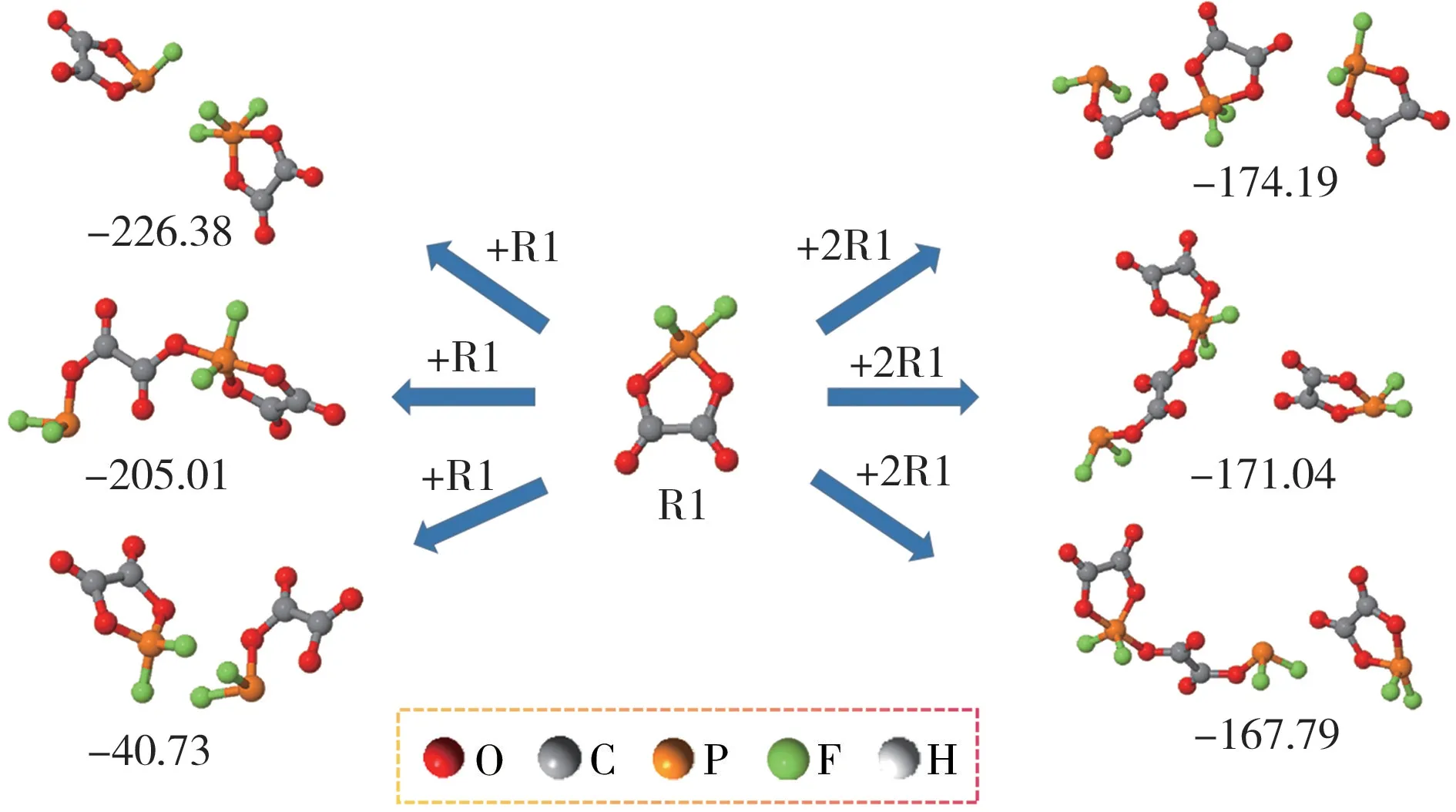
图8 R1 的聚合反应及相应的反应能(kJ/mol)Fig.8 Polymerization reaction of R1 and the corresponding reaction energy(kJ/mol)
R0 经过较低的能垒,生成含有R0 的产物M0;R1 则继续分解,生成产物M3。R1 具有较高的反应活性,可以与EC结合,可能会生成聚合物(见图7)。同时,从图8 可知,R1 的自由基聚合反应也能在电极表面形成界面膜,抑制电极液持续氧化反应。这与S.Jeong 等[14]的分析结果相一致,即Li-ODFP 会在锂离子电池正极表面生成含有磷酸盐的膜,这层氟代磷酸盐的低聚合物在抑制碳酸酯溶剂分子进一步氧化分解中起着关键的作用。
3 结论
本文作者采用量子化学计算方法,研究了常见锂盐的氧化稳定性和锂盐LiODFP 作为电解液成膜添加剂的作用机理。对单分子体系而言,ODFP-具有较高的氧化稳定性,低于无机锂盐阴离子PF6-和BF4-,但是比碳酸酯和其他有机锂盐阴离子(DFOB-、BOB-)更稳定。原因与ODFP-结构中含有有机环、吸电子基团的个数以及核心原子的半径大小有关。草酸根是LiODFP 中最不稳定的官能团,是发生氧化反应时电荷变化最大的部位。
另外,计算结果表明,所研究的体系氧化电位由高到低分别为:碳酸酯(包括EC、PC、DMC 和EMC)>ODFP-≈碳酸酯-ODFP-络合物,说明在电池充电过程中,电解液中的ODFP-阴离子将优于碳酸酯发生氧化反应。同时,与碳酸酯-PF6-、碳酸酯-BF4-络合物相比,碳酸酯-ODFP-发生氧化反应过程中没有HF 生成。研究EC+ODFP--e 的分解机理发现,ODFP-结构断裂分解的路径比EC 分子结构断裂分解所需能垒更低,表明ODFP-确实发生成膜反应。最容易进行(能垒最低)的反应路径是阴离子ODFP-结构开环,生成CO2和自由基R1。自由基R1 可进一步反应生成含有氟代磷酸盐单体的低聚物膜,这层聚合物膜有助于抑制碳酸酯基电解液直接与带电的正极表面接触。
