基于蒽核深蓝光材料的合成及电致发光性能
2024-03-11张越华邢龙江聂飞陈文铖穆英啸彭少洪何念孙宇曦曾庆明霍延平
张越华, 邢龙江, 聂飞, 陈文铖*, 穆英啸,彭少洪, 何念, 孙宇曦, 曾庆明, 霍延平,4*
(1. 广州城市职业学院, 广东 广州 510405; 2. 广东工业大学 轻工化工学院, 广东 广州 510006;3. 广东硕成科技股份有限公司, 广东 韶关 512600; 4. 广东工业大学 分析测试中心, 广东 广州 510006)
1 引言
有机发光二极管(Organic light-emitting diode, OLED)因其具有柔性显示、宽视角、响应快、效率高、超薄和功耗低等众多优势而成为近年来的研究热点[1-9]。OLED的光色由发光材料决定,高效稳定的蓝、绿、红三原色发光材料在全彩显示与白光照明领域具有重要地位。目前,绿光和红光材料在商用中已经取得了快速的发展;而蓝光材料,尤其是深蓝光材料,具有更宽的带隙,在器件工作中会由于高的能带壁障降低电子传输效率,使得器件的空穴和电子传输更加不平衡,从而导致器件的工作寿命变短。根据材料的发光的机制的不同,深蓝光材料主要分为三类:传统的荧光材料、磷光材料和热活化延迟荧光(Thermally activated delayed fluorescence, TADF)材料。在电致发光中,当电子和空穴注入器件中,会产生25%单线态和75%三线态激子,对于传统荧光材料,只能利用25%的单线态激子发出荧光,而75% 三线态激子通过非辐射途径损失。因此,传统荧光OLED的内量子效率(Internal quantum efficiency,IQE)上限为25%,一般来说,OLED的光输出效率在20%~30%之间,这就意味着传统荧光OLED的理论外量子效率(External quantum efficiency,EQE)范围仅为5%~7.5%[10-12]。而磷光材料含有重金属铂、铱等元素,通过自旋轨道耦合,使得25%的单线态激子通过系间窜越(Intersystem crossing, ISC)到三线态,使得器件的最大内量子效率达到100%。虽然器件的效率得到了提升,但是在高亮度下器件的效率滚降很严重,这是由于三线态激子浓度过高会引起三线态-三线态湮灭(Triplet-triplet annihilation, TTA)和三线态-单线态湮灭(Triplet-singlet annihilation, TSA)等,因此不能单独作为发光层使用,需要掺杂在合适的主体中[13-16]。此外,宽带隙磷光材料激发态能量高,金属配位键较共价键不稳定,导致其器件寿命短[17]。为了解决这类问题,研究者主要从分子设计和器件制备两方面入手,比如引入刚性配体和较强的金属-配体键,提升非辐射金属中心配体场态和通过梯度掺杂浓度使得激子复合区扩散到整个发光层,来抑制双分子湮灭效应。TADF材料是利用较小的单三态能级差,使得75%的三线态激子通过反向系间窜越(Reverse intersystem crossing,RISC)回到单线态实现近100%的内量子效率[18]。但是与磷光材料一样,TADF也面临在高亮度下的滚降严重问题,目前主要采取两种策略解决该问题。一方面,通过掺杂工艺来降低器件的效率滚降;另一方面,通过合理的分子设计,比如大扭曲分子内电荷转移态设计策略,通过引入大平面给受体单元,增大给受体二面角度,降低分子的前线分子轨道交叠来提高分子的反向系间窜越速率和增强分子内电荷转移(Intramolecular charge transfer, ICT)效应,但是强的ICT会导致光谱红移,很难实现深蓝光发射,因此在深蓝光材料的设计上存在着很大的挑战[19]。当下商业化的蓝光OLED仍然采用传统蓝色荧光材料,这主要是因为传统的荧光材料具有较长的器件工作寿命及高的色纯度,同时,传统荧光材料易通过分子结构设计调控材料性能。在蓝色磷光和热活化延迟荧光材料还未同时达到高效率和高的器件使用寿命时,传统蓝色荧光材料依然在蓝光OLED的制备中难以被取代。尽管如此,基于传统荧光材料的OLED的效率依然不尽人意。因此,开发高效稳定的深蓝光材料就成了OLED领域研究的重点和难点。
1998年,三线态-三线态湮灭(Triplet-triplet annihilation, TTA)首次被提出用于提高OLED的效率[20]。TTA需要两个单独的三线态(T1)激子相互作用,通过三线态融合得到一个单线态(S1)激子,S1激子再回到基态[17,21]。因此,基于TTA荧光材料的OLED的理论IQE由传统荧光OLED的25%提升到62.5%,包括来自本身S1态的25%和来自TTA的37.5%(75%×0.5)[22],且TTA材料可以不需要主体材料的参与来制备非掺杂OLED。另外,获得高的TTA效率需要满足2E(T1)>E(S1)。近年来,利用TTA机制提升器件的效率获得了很大的进展。2017年,Lu等合成了以菲并咪唑基团为弱电子给体、氰基为电子受体的化合物PIAnCN[23],并制备了非掺杂器件。测试结果表明,PIAnCN分子具有扭曲的分子结构,很大程度抑制了蒽核在固态下的π-π堆积,使得薄膜PLQY高达80%。化合物PIAnCN在1000 cd·m-2亮度下获得了9.44 %的最大EQE。通过瞬态电致测试发现延迟荧光,这种高效和低效率滚降的器件性能是因为TTA过程导致的。2019年,Wang等以—CN为吸电子基团、咔唑为给电子基团,合成了高效的深蓝光发光材料3CzAnBzt和pCzAnBzt[24]。这两个化合物制备的非掺杂器件的最大EQE分别为10.06%和9.23%,在1000 cd·m-2的亮度下,器件的EQE仍可以维持在8.97%和7.10%,CIE坐标分别是(0.14, 0.14)和(0.14, 0.10)。2022年,Promarak等在蒽的9,10号位引入给电子基团咔唑和吸电子基团四苯基咪唑设计合成了深蓝光材料HO-PIAC和PIAC,两个化合物的电致发射峰分别是440 nm和437 nm,制备的掺杂在主体材料CBP中的器件最大EQE分别是6.43%和4.51%,CIE坐标分别(0.15, 0.08)和(0.15, 0.07),实现了深蓝光发射。分子HO-PIAC的优异EL性能归因于分子内氢键的存在,赋予了更好的空穴传输能力和更大的TTA贡献[25]。2023年,Su等报道了具有TTA效应的化合物1NIAN和3NIAN[26]。采用两个化合物制备的非掺杂器件最大EQE分别达到10.9%和11.2%,CIE坐标分别为(0.15, 0.14)和(0.15, 0.15)。这是迄今为止报道的最高效率的蒽基TTA型深蓝光发射材料。理论和实验结果表明,借助有效的自旋轨道相互作用,即使三线态激子对的能级高于计算的第二三线激发态能级,TTA过程也可以得到进一步改善。相比于含重金属的磷光材料和TADF材料,TTA材料可以制备非掺杂器件,降低了器件制备工艺的复杂性;但是,目前报道的TTA最大的EQE效率仅为12%,如何尽最大限度利用TTA改善器件效率在分子设计上依然面临着挑战[27]。
蒽具有稳定高效的蓝光发射、合适的T1能级、稳定的电化学性能和高的PLQY等特点[23-24],是用来构建高效TTA型蓝光材料的常用基元。然而蒽的刚性平面结构容易导致聚集态下强的π-π相互作用,进而导致聚集态下的弱发射。通过相关基团的修饰能改善这个问题,研究人员主要对蒽的9和10位进行修饰。另外,引入给电子和吸电子基团也是常用的分子设计策略,能促进器件的载流子平衡。基于以上考虑,本工作在蒽的9和10位分别引入弱给电子基团二苯并噻吩和弱吸电子基团苯氰,设计合成了两个给体-受体(D-A)型深蓝光TTA材料2和3。其中,位阻基团二苯并噻吩的引入抑制了固态下分子的π-π堆积,引入弱的给电子和吸电子基团能抑制分子的ICT效应,进而实现深蓝光发射。我们对化合物2和3的热稳定性、电化学性质、光物理性质及电致发光性质进行了表征。
2 实验
2.1 仪器
核磁测试采用瑞士Bruker生产的AVANCEⅢ HD 400型核磁共振仪;高分辨质谱采用超高分辨四极杆组合静电场轨道阱液质联用仪;TGA测试采用STA449F5热重分析仪;电化学测试采用PGSTAT 302N电化学工作站;紫外可见吸收光谱采用UV-2700紫外可见分光光度计;荧光和磷光光谱、PLQY、寿命的测试采用FLS980荧光光谱仪。
2.2 合成
中间体4-(10-溴蒽-9-基)苯腈(1):将9, 10-二溴蒽(3.34 g, 10 mmol)、4-氰基苯硼酸(1.48 g, 10 mmol)、四(三苯基膦)钯(230 mg, 0.2 mmol)、碳酸钾(6.62 g, 48 mmol)和140 mL甲苯/乙醇/水(V∶V∶V=4∶2∶1)依次加入到250 mL两颈烧瓶中,将烧瓶在真空下抽空并在干燥氮气中置换三次,在120 ℃下加热回流搅拌避光反应12 h。使用饱和食盐水和二氯甲烷萃取,减压蒸馏,获得白色固体,使用硅胶粉作固定相,以石油醚/二氯甲烷为洗脱剂(4∶1,v/v),通过柱层析法获得1.78 g白色粉末(产率50%)。1H NMR(400 MHz, Chloroformd)δ8.64(dt,J=8.8, 1.0 Hz, 2H), 7.93~7.87(m,2H), 7.62(ddd,J=9.0, 6.4, 1.3 Hz, 2H), 7.52(ddd,J=19.2, 8.6, 1.3 Hz, 4H), 7.42(ddd,J=8.9, 6.4, 1.2 Hz, 2H)。
化合物4-(10-(二苯并[b,d]噻吩-4-基)蒽-9-基)苯腈(2):将中间体1(1.78 g, 5 mmol)、二苯并[b,d]噻吩-4-基硼酸(1.71 g, 7.5 mmol)、四(三苯基膦)钯(288 mg, 0.25 mmol)、碳酸钾(5.51 g, 40 mmol)和60 mL甲苯/乙醇/水(V∶V∶V=2∶1∶1)加入到100 mL两颈烧瓶中,将烧瓶在真空下抽空并在干燥氮气中置换三次。在120 ℃下加热回流搅拌,避光反应12 h。使用饱和食盐水和二氯甲烷萃取,减压蒸馏, 获得黄色固体, 使用硅胶粉作固定相,以石油醚/二氯甲烷为洗脱剂(2∶1,v/v),通过柱层析法获得1.61 g浅黄色粉末(产率70%)。1H NMR(400 MHz, Chloroform-d)δ8.39(dd,J=8.0, 1.1 Hz, 1H), 8.31(dd,J=7.9, 1.1 Hz, 1H),8.01~7.92(m, 2H), 7.79~7.70(m, 2H), 7.70~7.57(m, 7H), 7.51(td,J=7.6, 1.2 Hz, 1H), 7.47~7.37(m, 3H), 7.33(ddd,J=8.1, 6.5, 1.3 Hz, 2H)。13C NMR(101 MHz, Chloroform-d)δ144.40, 141.70,139.89, 136.08, 135.73, 135.61, 133.52, 132.37,132.34, 132.30, 129.64, 129.47, 129.45, 127.00,126.68, 126.35, 126.00, 125.79, 125.03, 124.58,122.87, 121.95, 121.19, 118.92, 111.79, 77.41,77.09, 76.77。HRMS(M)m/z: 462.13017[M+H]+(calcd: 462.13165)。
化合物4-(10-(二苯并[b,d]噻吩-2-基)蒽-9-基)苯腈(3):化合物3的合成步骤与化合物2类似,将原料二苯并[b, d]噻吩-4-基硼酸换成二苯并[b, d]噻吩-2-基硼酸,通过柱层析法获得1.44 g浅黄色粉末(产率63%)。1H NMR(400 MHz, Chloroformd)δ8.28(d,J=1.6 Hz, 1H), 8.11(dd, J=8.1,1.8 Hz, 2H), 8.01~7.90(m, 3H), 7.83~7.75(m, 2H),7.70~7.64(m, 2H),7.63~7.55(m, 3H), 7.52(td,J=7.6, 1.3 Hz, 1H), 7.45(td,J=7.5, 1.2 Hz, 1H),7.38(dddd,J=16.4, 7.9, 6.4, 1.5 Hz, 4H)。13C NMR(101 MHz, Chloroform-d)δ144.52, 140.02,138.94, 137.92, 135.94, 135.37, 134.92, 134.78,132.36, 132.32, 130.18, 129.80, 129.46, 127.31,127.08, 126.10, 125.82, 125.41, 124.59, 124.24,123.03, 122.91, 121.78, 118.91, 111.73。HRMS(M)m/z: 462.13020 [M+H]+(calcd: 462.13165)。
3 结果与讨论
目标化合物2和3的合成路线如图1所示,通过核磁共振氢谱(1H NMR)、核磁共振碳谱(13C NMR)和高分辨质谱(HRMS)等表征手段对合成的中间体及终产物的化学结构进行鉴定。
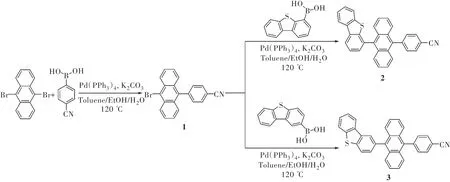
图1 化合物2和3的合成路线Fig.1 Synthetic route of compounds 2 and 3
3.1 热稳定性和电化学性质
利用真空蒸镀工艺制备OLED器件需要保证化合物具有良好的热稳定性。在氮气气氛下,我们对两个化合物进行了热重分析(Thermogravimetric analysis, TGA),如图2和表1所示,化合物2和3的热分解温度(Td,失重5%时对应的温度)分别为384 ℃和386 ℃,这表明两个化合物具有较高的热稳定性。我们通过循环伏安法测试的化合物2和3的氧化曲线如图3所示,两个化合物表现出可逆的氧化过程,表明它们具有较高的电化学稳定性。根据测得的氧化电位和公式EHOMO=-(Eonset,oxvsFc+ /Fc+ 4.8) eV,经计算得到化合物2和3的HOMO能级分别为-5.73 eV和-5.66 eV。通过化合物在纯膜中的紫外可见吸收光谱的起始吸收波长可计算出化合物2和3的HOMO-LUMO能隙Eg分别为2.90 eV和2.87 eV。由ELUMO=EHOMO+Eg可得到化合物2和3的LUMO能级分别为-2.83 eV和-2.79 eV。
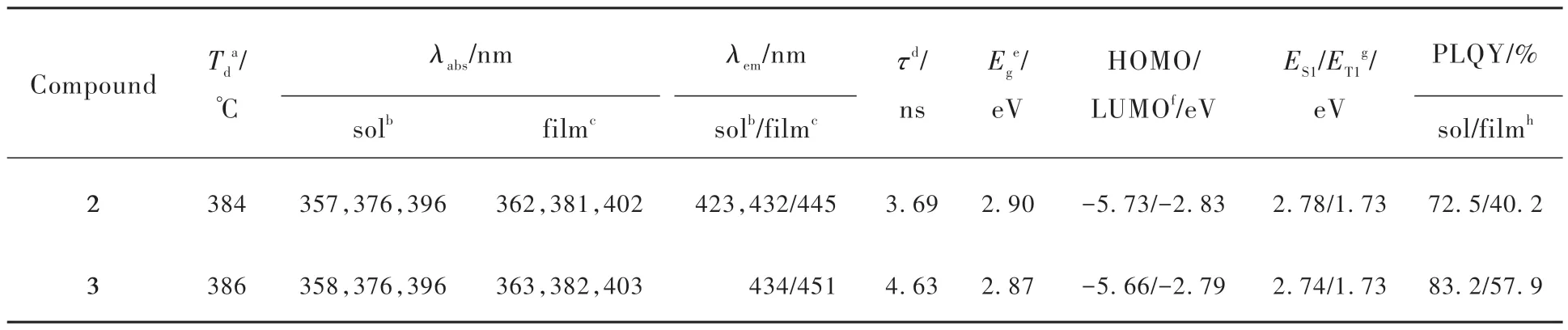
表1 化合物2和3的光物理数据汇总Tab.1 Summary of photophysical data of compounds 2 and 3

图2 化合物2和3的TGA曲线Fig.2 TGA curves of compounds 2 and 3

图3 化合物2和3的氧化曲线Fig.3 Oxidation curves of compounds 2 and 3
3.2 理论计算
为了评估化合物2和3的基态分子构型、最高占据分子轨道/最低未占据分子轨道(Highest occupied molecular orbital, HOMO/Lowest unoccupied molecular orbital, LUMO)能级、电子云分布和激发态能级,我们采用B3LYP/6-31G(d)基组对化合物进行了密度泛函理论(Density functional theory,DFT)和含时密度泛函理论(Time-dependent density functional theory, TD-DFT)计算。如图4所示,化合物2和3呈现出相似的电子云分布,HOMO和LUMO均主要分布在蒽核上,只有一小部分LUMO分布在苯腈上,这表明化合物2和3主要是局域激发态(Local excited, LE)性质。化合物2和3的HOMO能级分别为-5.41 eV和-5.40 eV,LUMO能级分别为-1.93 eV和-1.92 eV,Eg均为3.48 eV。化合物2的最低单重态(S1)和最低三重态(T1)能级分别为3.13 eV和1.73 eV,化合物3的分别为3.12 eV和1.73 eV。两个化合物的2E(T1)>E(S1),支持TTA过程的发生。
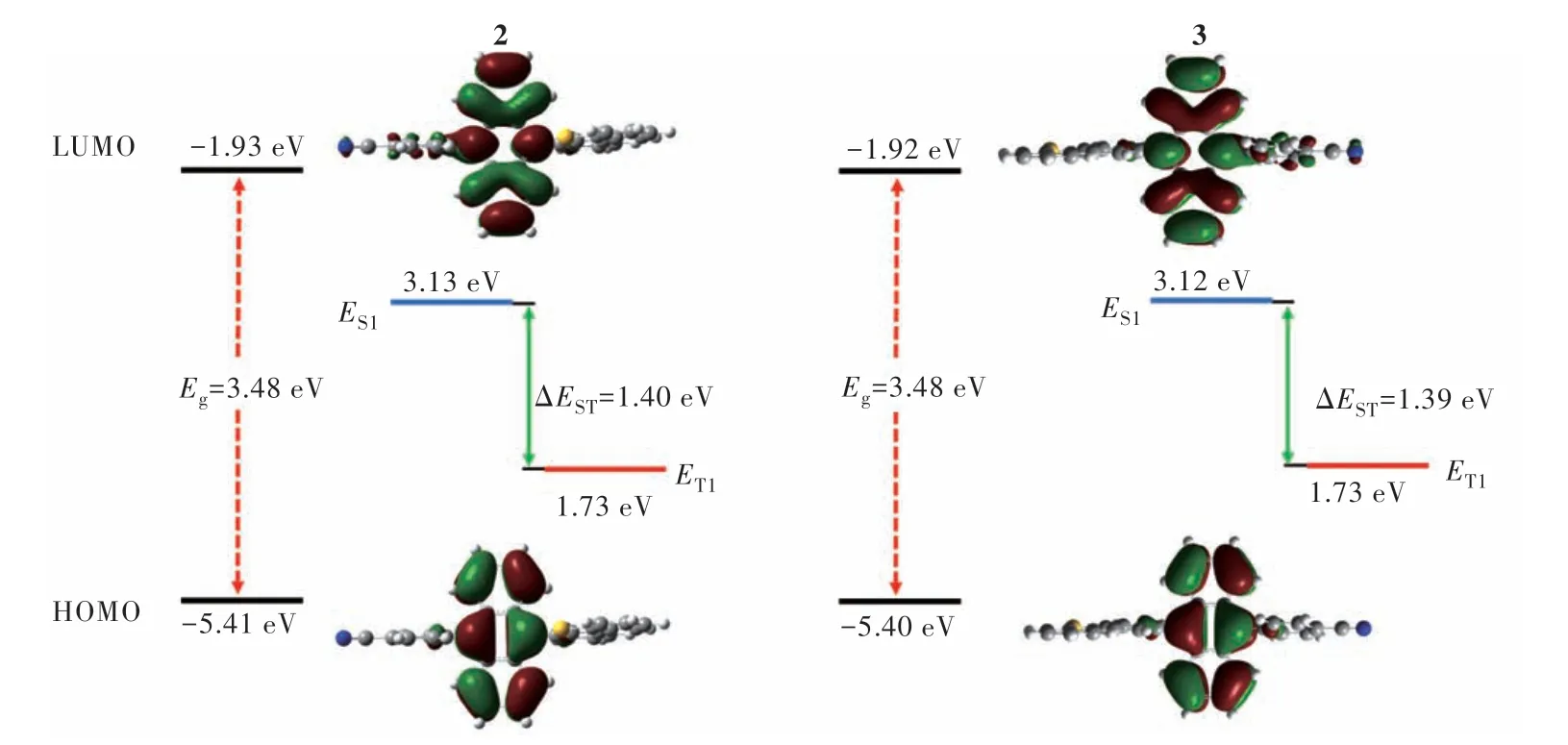
图4 优化的化合物2和3的分子几何结构及HOMO和LUMO电子云分布Fig.4 The optimized molecular geometries and HOMO and LUMO electron cloud distributions of compounds 2 and 3
3.3 光物理性质
化合物2和3在甲苯溶剂中的紫外可见吸收光谱图如图5(a)所示。两个化合物表现出相似的吸收带:350~400 nm,这可归属于蒽核的π-π*跃迁[26]。图5(a)、(b)的光致发光光谱中,化合物2在甲苯溶液中表现出了精细结构发射,发射峰分别为423 nm和432 nm;而化合物3只有一个发射峰,在434 nm处。这表明化合物2具有更加明显的局域激发的性质,这是因为化合物2的空间结构更加扭曲,导致分子的电荷转移(Charge transfer, CT)性质更弱。通过真空蒸镀工艺将化合物2和3蒸镀在石英基质上制备100 nm厚的非掺杂膜,化合物2和3在纯膜下的发射峰分别是445 nm和451 nm,相比在甲苯溶液中,光谱红移15 nm左右,这可归因于纯膜状态下分子间的紧密堆积使得分子间作用力增强。另外,我们测试了化合物2和3在甲苯溶剂和纯膜中的光致发光量子产率(Photoluminescence quantum yield, PLQY)以及纯膜状态下的荧光寿命(图6和表1)。两个化合物均显示出高效的PLQY,分别为72.5%/40.2%和83.2%/57.9%,并且荧光寿命均在纳秒级别(分别为3.69 ns和4.63 ns)。这些光物理特性表明,两个化合物用于制备深蓝光非掺杂OLED器件具有潜在的应用前景。
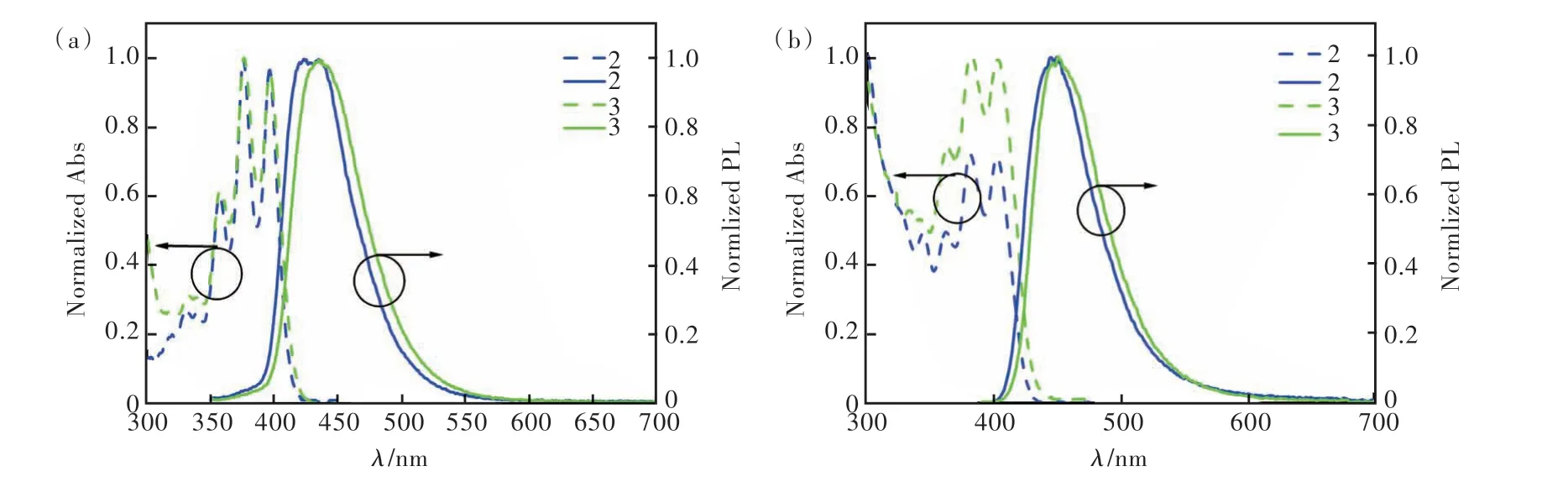
图5 化合物2和3在甲苯溶剂中(a)和纯膜中(b)的归一化紫外可见吸收光谱和光致发光光谱Fig.5 Normalized UV-Vis absorption spectra and photoluminescence spectra of compounds 2 and 3 in toluene solvent(a) and neat film(b)
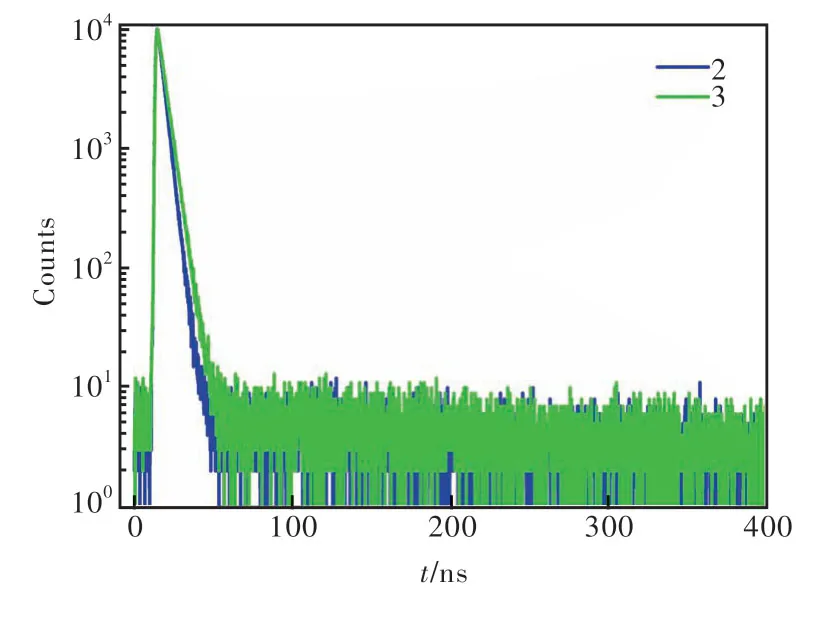
图6 化合物2和3的瞬态PL光谱Fig.6 Transient PL spectra of compounds 2 and 3
化合物2和3在不同极性溶剂中的光致发光光谱分别如图7(a)、(b)所示。从图中我们可以看出,两个化合物在不同极性溶剂中的发射光谱均无明显变化,说明两个化合物均表现出微弱的溶剂化效应,这表明两个化合物的S1态具有明显的LE态特征,而CT态特征较微弱,与理论计算的结果相吻合。化合物2和3以2%掺杂在聚甲基丙烯酸甲酯(PMMA)/二苯甲酮=4/1基质中,在室温下测得的磷光光谱和寿命如图8所示。两个化合物的磷光光谱均表现出明显精细振动结构,表明它们的T1态具有LE态特征。光谱的0-0跃迁位于715 nm 处,计算得出T1能级为1.73 eV,这与文献报道的蒽衍生物的T1能级相似[28]。根据前文两个化合物在纯膜中的发射波长计算得到它们的S1能级分别为2.78 eV和2.74 eV,因此,满足2E(T1)>E(S1),与理论计算相符。另外,我们测得化合物2和3在715 nm处的寿命分别为9.4 ms和7.9 ms。
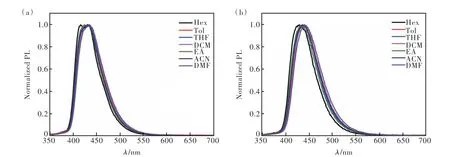
图7 化合物2(a)和3(b)在不同溶剂中的光致发光光谱Fig.7 Photoluminescence spectra of compounds 2(a) and 3(b) in different solvents
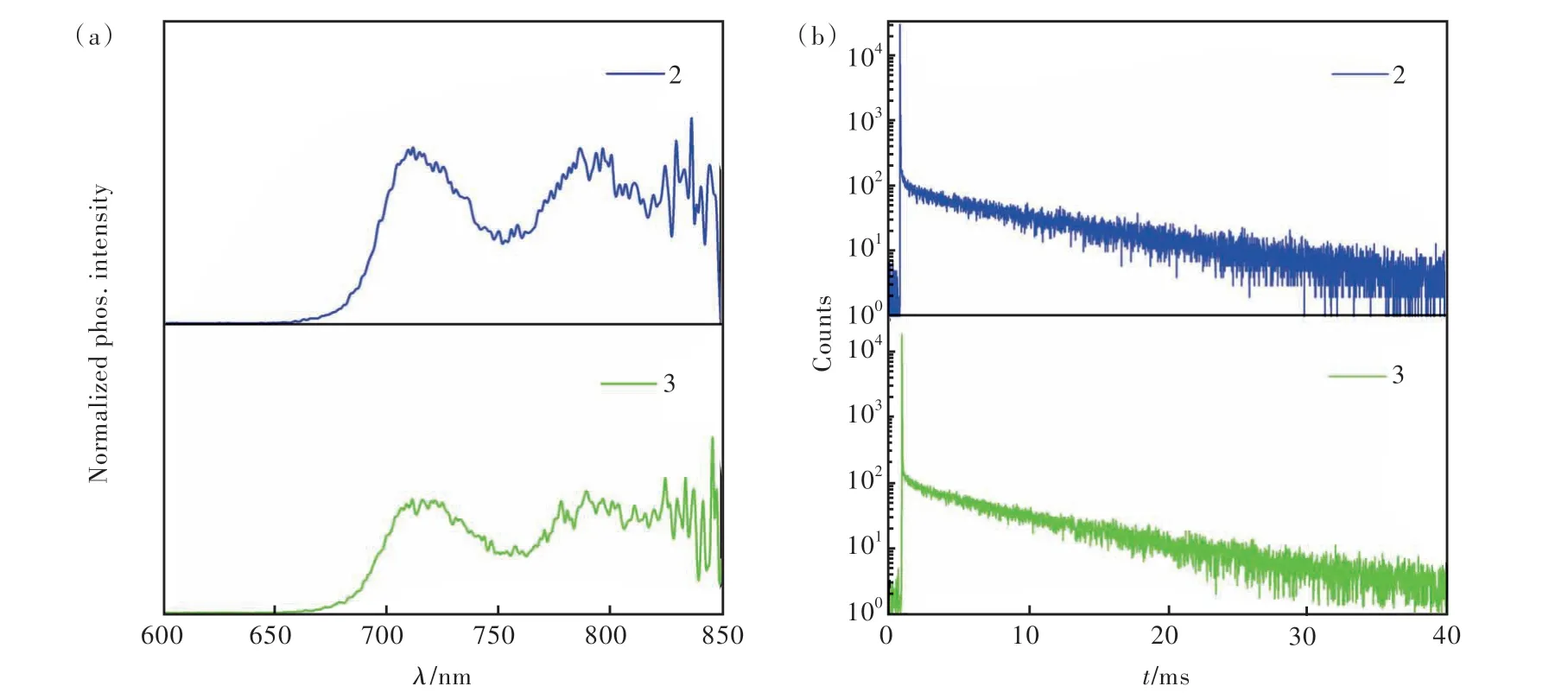
图8 以2%掺杂在PMMA/二苯甲酮=4/1基质中得到的化合物2和3延迟5 ms的磷光光谱(a)和瞬态PL光谱(b)Fig.8 Compounds 2 and 3 doped with 2% PMMA/benzophenone=4/1 have phosphorescent spectra with 5 ms delay(a) and transient PL spectra(b)
为了验证两个分子具有TTA性质,我们进行了三重态-三重态湮灭上转换(Triplet-triplet annihilation upconversion, TTA-UC)测试,引入光敏剂八乙基卟啉铂(Ⅱ)(PtOEP)。当光敏剂在532 nm的激光激发后,由于铂的“重原子效应”,S1态激子通过ISC到T1态,光敏剂的T1态的能量经过Dexter途径转移到受体的T1态,此后受体的T1态激子相互碰撞会产生上转换荧光。Monguzzi等已经证明,通过TTA过程产生的上转换荧光强度是遵循独特的功率依赖性,上转换荧光强度随激光功率变化呈现出慢TTA区域(在低激发功率)和快TTA区域(在高激发功率)[29]。如图S8(SI)所示,化合物2和3在低激光功率下,它们的斜率几乎是二次的(分别为1.93和1.77);然而,在高激光功率下,斜率分别为1.25 和1.09,几乎是线性相关的。因此,利用上转换荧光随功率变化直接证明了这两个发光材料均具有TTA过程。
3.4 电致发光性能
我们将化合物2和3作为发光层制作了结构为ITO/HATCN (5 nm)/TAPC (40 nm)/TCTA (5 nm)/2 or 3 (20 nm)/TmPyPB (40 nm)/LiF (1 nm)/Al的非掺杂深蓝光OLED器件A和B(图9(a))。其中,2,3,6,7,10,11-六氰基-1,4,5,8,9,12-六氮杂苯并菲(HATCN)、4,4'-环己基二[N,N-二(4-甲基苯基)苯胺](TAPC)、4,4',4″-三(咔唑-9-基)三苯胺(TCTA)、3,3'-[5'-[3-(3-吡啶基)苯基][1,1'∶3',1″-三联苯]-3,3″-二基]二吡啶(TmPyPB)、LiF分别用作器件的空穴注入层、空穴传输层、电子阻挡层、电子传输层和电子注入层。器件中所用材料的化学结构如图9(b)所示。两个器件的电致发光数据汇总于表2中。器件A和B在1000 cd·m-2亮度下的电致发光光谱如图10(a)所示,两个器件均呈现深蓝光发射,发射峰分别位于448 nm和458 nm处,半峰宽分别是77 nm和75 nm。器件的电致发光光谱与相应化合物在纯膜中的光致发光光谱相似,表明激子的复合主要发生在发光层中。两个器件在1000 cd·m-2亮度下的CIE色坐标分别为(0.15,0.12)和(0.15, 0.14),接近蓝光标准CIE色坐标(0.14, 0.08)。两个器件的启动电压(Turn-on voltage,Von)较低,不超过4.0 V(图10(b))。基于化合物2和3的器件A和B的最大电流效率(Current efficiency, CE)分别为4.2 cd·A-1和6.9 cd·A-1, 最大功率效率(Power efficiency, PE)分别为2.3 lm·W-1和3.6 lm·W-1,EQE分别为3.8%和5.6%。在1000 cd·m-2亮度下,两个器件的CE分别为4.16.7 cd·A-1和6.7 cd·A-1,PE分别为1.7 lm·W-1和2.8 lm·W-1,EQE分别为3.7%和5.4%。值得注意的是,化合物2 的最大 EQE明显低于化合物3,根据评估器件的EQE公式:
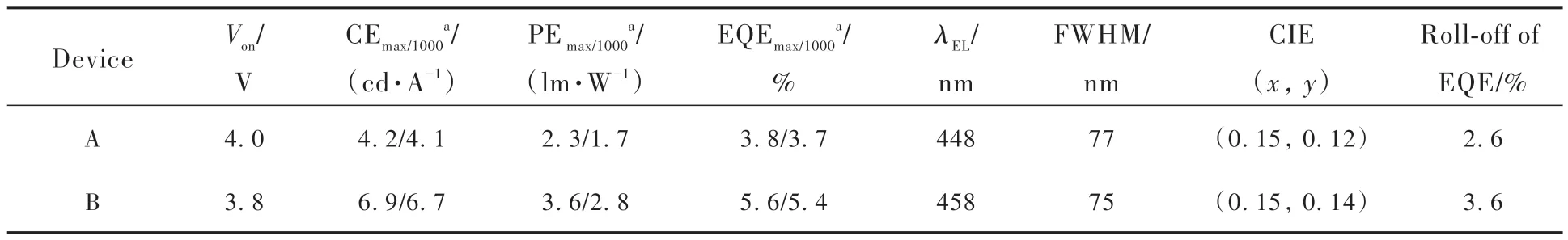
表2 基于化合物2和3的非掺杂器件A和B的电致发光数据Tab.2 Electroluminescence data of undoped devices A and B based on compounds 2 and 3
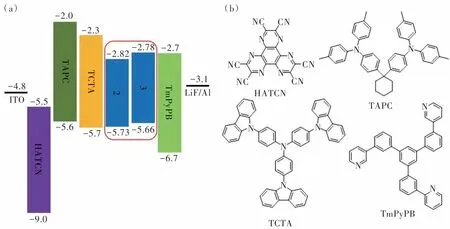
图9 非掺杂器件的能级图(a)及器件中所用材料的化学结构(b)Fig.9 Energy level diagram of undoped devices(a) and chemical structure of materials used in devices(b)
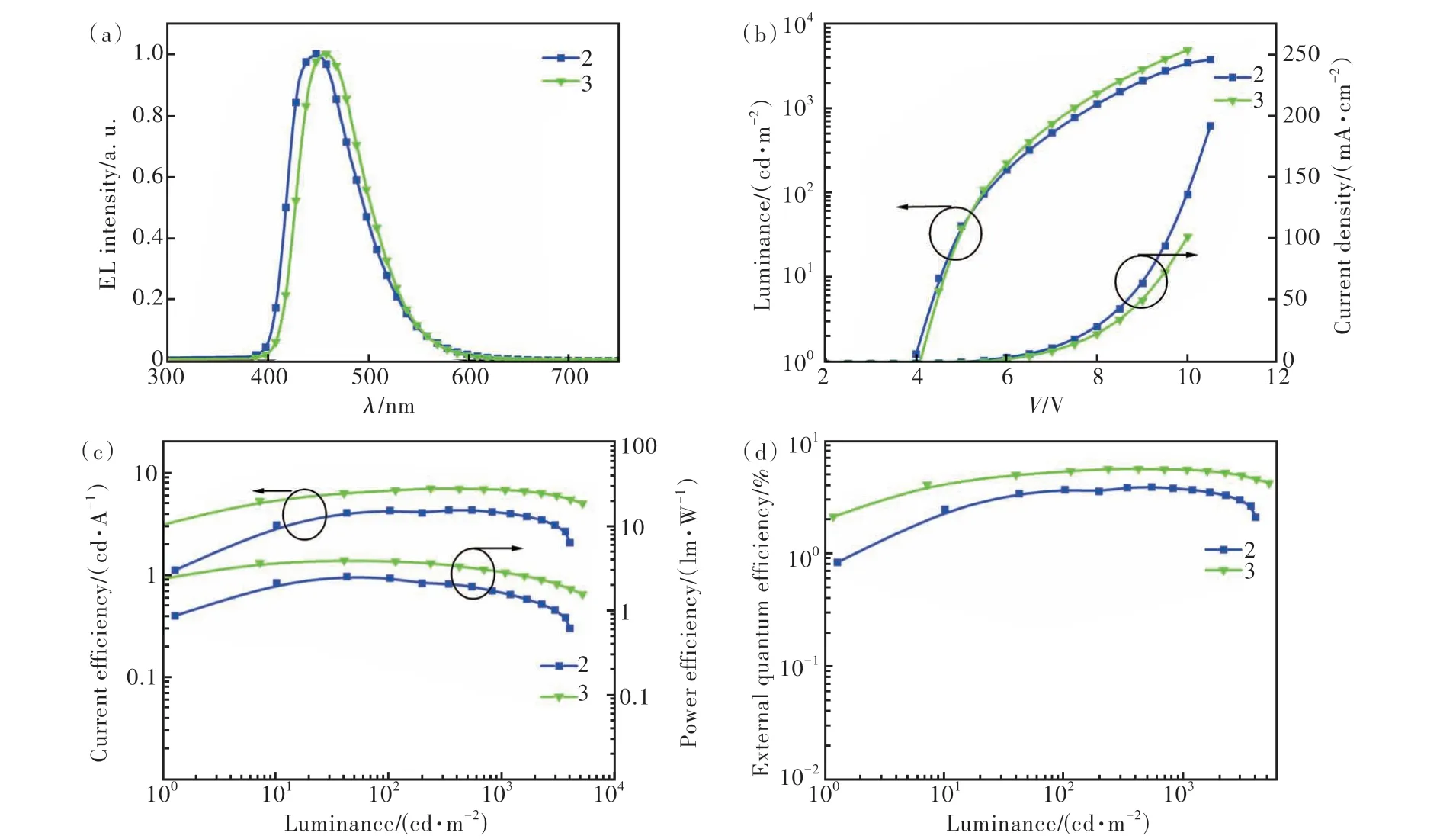
图10 器件的电致发光光谱(a)、电流密度-电压-亮度(J-V-L)曲线(b)、电流效率-亮度-功率效率(CE-L-PE)曲线(c)、外量子效率-亮度(EQE-L)曲线(d)Fig.10 The electroluminescence spectra(a),current density-voltage-luminance(J-V-L) curves(b),current efficiency-luminance-power efficiency(CE-L-PE) curves(c), external quantum efficiency-luminance(EQE-L) curves(d) of the device
其中,γ是理想情况下100%的空穴和电子复合效率,ΦPL代表薄膜状态下的PLQY,ηr表示辐射激子产生的单重态激子(对于传统荧光材料只有25%),ηop代表光输出耦合因子(0.2~0.3)。化合物的荧光量子效率会影响器件的效率,因此主要原因是前者的荧光量子效率要低于化合物3。当假设ηop为0.3时,通过计算我们得到化合物2和3 的ηr分别是31.6%和32.2%,再结合化合物较大的ΔEST和TTA-UC测试,证明了是TTA效应贡献器件的EQE。如表2所示,基于化合物2和3的器件A和B的EQE从最大值下降到1000 cd·m-2亮度下的值,效率滚降分别为2.6%和3.6%,表现出极低的效率滚降。较低的Von和极低的效率滚降表明发光材料与各功能层材料能级之间匹配度高,电子和空穴能更好地注入与传输。
4 结论
本文以蒽单元作为蓝光TTA发光核,通过在蒽的9和10位分别引入弱给电子基团二苯并噻吩和弱吸电子基团苯氰,设计合成了两个D-A型深蓝光TTA材料2和3,并对它们的热稳定性、电化学性质、光物理性质及电致发光性质进行了系统的表征与分析。二苯并噻吩的引入能增大分子的空间位阻,抑制大平面蒽核在固态下的π-π堆积。引入相对较弱的给电子和吸电子基团能抑制分子的ICT效应导致的发光红移,有利于实现深蓝光发射。两个化合物具有较高的热稳定性,其热分解温度分别为384 ℃和386 ℃,有利于蒸镀器件的制备。两个化合物在甲苯溶剂和纯膜中均表现出深蓝光发射,其中纯膜下的发射峰分别位于445 nm和451 nm处。两个化合物均表现出较高的纯膜PLQY(40.2%和57.9%)。基于化合物2和3的非掺杂OLED器件的电致发射波长分别位于448 nm和458 nm处。两个器件的最大CE分别为4.2 cd·A-1和6.9 cd·A-1,最大PE分别为2.3 lm·W-1和3.6 lm·W-1,最大EQE分别为3.8%和5.6%。在1000 cd·m-2亮度下,器件的CE分别为4.1 cd·A-1和6.7 cd·A-1,PE分别为1.7 lm·W-1和2.8 lm·W-1,EQE分别为3.7%和5.4%。可见,两个器件均表现出较好的发光效率,且拥有极低的效率滚降(2.0%和3.5%)。另外, 我们注意到化合物2和3仅是二苯并噻吩的取代位点不同,而基于化合物3的器件具有更好的发光效率,但基于化合物2的器件具有更深的蓝色荧光。
本文补充文件和专家审稿意见及作者回复内容的下载地址:http://cjl.lightpublishing.cn/thesisDetails#10.37188/CJL.20230288.
