土壤中有机质镉的同位素组成特征:来自第一性原理计算的约束
2024-03-09张田迪邢乐才赵浩男谷一帆何洪涛
张田迪,邢乐才,2,赵浩男,李 英,谷一帆,杨 阳,何洪涛,2*
(1.河北工程大学 地球科学与工程学院,河北 邯郸 056038;2.河北工程大学 河北省资源调查与研究重点实验室,河北 邯郸 056038)
镉(Cd)是银白色带蓝色光泽的金属元素,位于周期表的第五周期第二副族中。它的偶次原子结构决定了它易失掉最外层电子,成为对称结构的钯型稳定离子[1]。因此,在表生环境下Cd主要以+2价形式存在。重金属Cd主要通过铅锌矿石的开采和冶炼、工业制造和滥用化学肥料等人为活动排放到土壤中,已成为全球土壤主要重金属污染物之一[2]。大多数地质及环境样品中Cd含量极低,痕量、超痕量Cd元素含量的准确测定及高精度的同位素分析仍是一项具有挑战性的研究工作[3]。然而,当环境中的某些物理化学过程能够导致较大的金属同位素分馏时,示踪工作变得复杂,需要识别和量化单个过程中的同位素分馏程度。例如,有机质表面吸附及螯合过程在表生条件下普遍存在,是影响Cd同位素分馏的重要过程,但该过程中的同位素分馏尚未得到有效的限定。有机质作为土壤的重要组成部分,占土壤固体部分的5%。而有机质中85%是腐殖质。腐殖质作为环境中许多必需和有毒微量元素的吸附剂,在控制其在土壤、沉积物和水生系统中的流动性和生物有效性中发挥着重要的作用[4]。
确定分馏特征(程度)的方法主要有三种,分别是理论计算、实验测定、野外半定量评估[5]。迄今为止,只有少数实验室对吸附和络合过程中镉同位素分馏进行了量化[6]。然而,同位素交换平衡所需的时间尺度不同于元素平衡,很难判断同位素交换反应是否达到真正意义上的平衡。因此,将实验结果外推到自然条件时必须谨慎。如今,基于密度泛函数理论(DFT)的第一性原理计算方法已成为实验研究的重要补充[7]。它可以提供同位素交换反应中关键步骤的详细信息。该技术的主要原理是分子的各种热力学性质(反应吉布斯自由能变化、IR活性、化学位移等)与其电子密度具有紧密的函数关系。通过求解分子的Schrödinger方程,可以得到对应于最稳定的化学结构的电子能和体系波函数。作为同位素分馏因子计算中的关键参数,谐波振动频率即为电子能对原子坐标的二阶偏导。
1 理论与方法
本研究采用第一性原理计算方法对分子团簇进行几何构型优化。使用BP86作为交换相关函数,采用混合基组描述体系的波函数,即Cd选择LanL2MB赝势基组[8],H,O,N, 和 S原子选择6-311G(d)全电子基组[9]。几何优化完成后,我们检查每一组构型的谐振动频率中是否存在虚频,以确认该构型是否对应势能面上的局部极小值。体系电子能和谐振动频率计算均通过Gaussian09/ Gaussian16软件[10-11]完成。
1.1 平衡分馏系数计算
Urey 等[12-13]描述了化合物和单原子理想气体之间同位素交换的场景。他们提出了一个重要的参数,即约化配分函数(Reduced Partition Function Ratio, RPFR),来量化化合物相对富集重同位素的程度。该参数值大小主要取决于同位素替换前后的化合物的谐振动频率。RPFR值越大于1,表明该物质中的重同位素富集程度越大。
对于化合物和单原子理想气体之间的同位素交换反应:
AX+X'AX'+X
(1)
式中,X'代表含有重同位素的单原子理想气体,X代表含有轻同位素的单原子理想气体,AX代表具有轻同位素的化合物,AX'代表具有重同位素的化合物。该反应的平衡常数可表示为

(2)
其中Qtrans、Qrot、Qvib、Qelec分别表示平动配分函数、转动配分函数、振动配分函数、电子配分函数。对于轻元素来说,同位素替换前后的化合物的基态电子能差异基本上是无法区分的,因此可以忽略电子配分函数对RPFR值的贡献。具体而言,平动配分函数、转动配分函数和振动配分函数可以表示成:
(3)
(4)

(5)
其中V是分子体积,M是分子质量,IA是转动轴A的转动惯量,σ是分子的对称数。ui=hcωi/kT,h为普朗克常数,k为玻尔兹曼常数,T为绝对温度(K),c是光速,ω为谐振动频率(cm-1)。使用Teller-Redlich[14]公式进一步近似,可以得到:

(6)
最终,平动配分函数比和转动配分函数比可以使用振动频率来表示。通过一系列数学变换,约化配分函数比(RPFR(AX/AX’))定义如下:

(7)
RPFR值等价于化学领域常用的β因子,即化合物与单原子理想气体的同位素分馏因子[15]。当平衡共存的A、B两相的RPFR已知时,则同位素分馏系数α可以由它们的RPFR(或β因子)的比值来表示:

(8)
两相之间的同位素平衡分馏值由公式:ΔA-B=δA-δB≈1 000(lnβA-lnβB)=1 000lnαA-B计算得到。该公式将同位素理论计算与实验测定工作联系起来,便于同位素分馏数据的对比。
同时,本文利用最小均方根偏差法[16]来确定每个含Cd物种RPFR值的平台值:
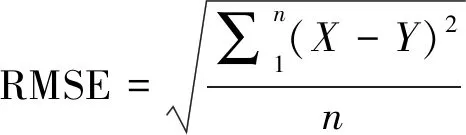
(9)
其中X代表我们计算出的每一个RPFR值,Y为预期的平台值。RMSE值随Y值的变化而变化,RMSE值取得最小值时对应的Y值,即为某含Cd物种的平台值。
1.2 分子簇模型构建
腐殖酸的具体结构尚不清楚且分子量较大,其复杂程度超过了现阶段的计算模拟能力,但是已知腐殖酸的各吸附点位上能够络合金属离子的结构主要为含氧官能团,如羧基和酚羟基等。因此,我们使用小分子有机酸来代替腐殖酸,以此构建OSCs和MOFs的初始构型。OSCs初始构型优化结束后,在优化好的构型上继续添加水分子,搭建下一组吸附态的初始构型。每次加6~7个水分子,建立下一组吸附Cd的初始构型。添加的水分子放置在氢键的成键范围内(1.8~2.1 Å),并且使水分子中的氧原子(或者氢原子)尽可能与其他水分子的氢原子(或者氧原子)形成氢键。每完成一组构型优化,提取谐振动频率,计算RPFR值。随着添加水分子数量的增加,RPFR值逐渐收敛于平台值。同时,腐殖酸中的N、S也可作为供体原子与金属络合。根据CCDC晶体数据库中心提供的构型数据,对其构型进行修改来构建MOFs构型。OSCs和MOFs之间的区别在于,OSCs构型模拟Cd吸附在有机质表面,周围有较多的水分子,而MOFs构型模拟Cd脱水进入有机质内部,配体中无水或水分子较少。在MOFs构型中我们还考虑了具有羟基、羧基、N原子和S原子不同组合的有机配体情形。
2 结果与讨论
2.1 OSCs和MOFs的几何构型
优化完成的OSCs和MOFs构型见图1。同位素平衡分馏主要受到局部结构的控制[17],一般为距离目标原子3~5 Å范围内的化学作用。因此,对Cd原子周围化学环境模拟的准确性主要决定了同位素分馏系数计算的准确性。从图1、图2中,可以看出OSCs和MOFs均保留住了Cd-O八面体的初始构型。在OSCs中Cd-O平均键长为2.33 Å,在MOFs中Cd-O平均键长为2.40 Å。同时,Cd与N或S配位,将会增大Cd与第一配位层的原子间距。计算的Cd-O键长与公开发表的数据(表1)吻合较好,在一定程度上保证了1 000 lnβ的正确性。

图1 Cd OSCs几何构型

表1 含Cd 物种与水溶液之间的平衡同位素分馏

图2 Cd MOFs几何构型
2.2 同位素平衡分馏
基于水溶液的RPFR值(1.002 887)[7],计算出25 ℃下OSCs和MOFs与水溶液之间的Cd同位素平衡分馏值(图3)。发现,OSCs溶剂效应模拟的准确性直接影响到Cd同位素平衡分馏值的选取。随着水化层中水分子数的逐渐增加,分馏值由波动状态逐渐收敛,直到达到平台值。

图3 溶剂效应对OSCs与溶液间Cd同位素分馏的影响
最终,获得了OCSs与溶液之间的Cd同位素平衡分馏为-0.34‰~0.02‰,最大值为0.02‰,最小值为-0.34‰,平均值为-0.17‰。同样地,我们获得了MOFs与溶液之间的Cd同位素平衡分馏为-1.92‰~-0.29‰,最大值为-0.29‰,最小值为-1.92‰,平均值为-0.87‰。明显地,当Cd与N或S配位,同位素分馏更为显著。
计算结果与土壤剖面的测试结果是一致的(图3)。由于土壤矿物表面吸附过程产生的Cd同位素分馏在分馏方向和分馏程度上与有机质表面吸附过程相当,其中铁(氢氧)氧化物分馏值为(-0.53±0.04)‰[25],锰氢氧化物分馏值为(-0.24±0.12)‰~(-0.54±0.14)‰[26],粘土矿物分馏值为-0.13‰[22],这使得土壤体系Cd同位素组成的解释变得复杂。
目前,可用的有机质与水溶液之间的Cd同位素分馏只有Zhao等人[21]的计算和Ratié等人[6]的实验。Zhao等人[21]计算的Cd同位素分馏为正值,表明有机质更倾向富集重Cd同位素,这与我们的结果相反(图4),这是因为他们的计算中使用的是隐式溶剂模型,而不是显示溶剂模型。这可能导致Cd原子周围局部结构的不精确表达,这对同位素平衡分馏计算是极为重要的。Ratié等[27]认为当Cd与腐殖酸的羧基位点形成配合物时,Cd同位素分馏为(-0.15±0.01)‰,这与我们关于乙酸和苯甲酸体系的计算是一致的。

图4 含Cd有机物-溶液之间同位素平衡分馏

3 结论
25 ℃以下,有机质表面配合物(OSCs)与水溶液之间的Cd同位素平衡分馏较小且不敏感,即Δ114/110CdOSCs-aq=-0.34‰~0.02‰。相比之下,金属-有机框架(MOFs)的形成产生了显著的Cd同位素分馏,即Δ114/110CdMOFs-aq=-1.92‰~}。当将OSCs和MOFs视为两个端元时,OSCs和MOFs信号的二元混合模型可以合理地解释已发表的有机质土壤的Cd同位素组成测定结果。
