基于肠道微生物学及代谢组学研究2型糖尿病患者肠道菌群结构及血清代谢产物的特征
2024-03-09阿孜古丽买合买提米日古丽吾木哈斯木李雪梅塞娜瓦尔阿西木穆叶斯尔哈斯木
阿孜古丽·买合买提, 米日古丽·吾木哈斯木, 李雪梅, 塞娜瓦尔·阿西木, 穆叶斯尔·哈斯木△
新疆维吾尔自治区妇幼保健院 1呼吸内分泌科, 2冠心病肾病科(新疆乌鲁木齐 830000)
糖尿病(DM)作为以脂肪、蛋白质、水、电解质代谢紊乱的一组代谢性综合征,调查表明[1],我国糖尿病发生率已经从1980年的0.67%升高至2018年的10.9%,患病人数已是全球首位,且发病年轻化趋势越加明显。近年来随着分子生物学技术的不断发展,人类对肠道微生物的认知有了巨大的进步,肠道菌群与疾病的相关性研究已经成为目前关注的热点。相关研究显示[2],肠道菌群与宿主的消化、代谢、免疫等方面密切相关,被许多学者比喻为人类的一个额外“器官”。相关报道表明[3],肠道菌群不但可以从正常食物中获得额外能量促进肥胖的发生,还能够通过分泌内毒素导致机体发生内毒素血症,同时还可调节组织生物活性的脂肪酸合成等一系列过程参与DM等代谢性疾病的发生、发展。在另一领域,代谢组学是一类研究代谢性疾病的重要科学,通过研究机体代谢组学的特征谱能够帮助人们理解疾病发生发展的机制以及获得相应的生物学标志物。20世纪末国外有学者就对糖尿病的代谢谱进行分析,得出DM患者与正常人代谢谱之间存在较为明显的差异[4]。国外有研究发现[5],通过LC-MS和二维GC-MS对儿童血清脂质谱、氨基酸和其他小分子代谢物进行分析,发现肠道菌群与糖尿病的发生有关。本研究拟通过分析2型糖尿病(T2DM)患者的肠道菌群结构多样性特征和代谢组学差异,为临床实践提供参考依据,报告如下。
1 资料与方法
1.1 纳入与排除标准 纳入2021年6月至2022年12月新疆维吾尔自治区妇幼保健院就诊的初发T2DM患者100例,设为DM组;另同期招募健康志愿者100例,设为对照组。年龄20~65岁,同意提供粪便和血清标本,询问相应病史症状,完善相关检查,研究对象均自愿签署由我院伦理委员会批准的知情同意书及相关文件。研究经新疆维吾尔自治区妇幼保健院伦理委员会审批通过(K-2021052)。纳入标准:(1)病例组:首次诊断为T2DM患者[6];年龄20~65岁;符合其他各项健康体检指标;(2)对照组:排除糖尿病的健康志愿者;年龄、性别、民族与病例组相匹配;符合各项健康体检指标。排除标准:(1)血液系统,中枢神经系统疾病、风湿活动、自身免疫性疾病;(2)急、慢性消化道感染,慢性腹泻、便秘、消化道溃疡活动期及愈合期、炎症性肠病、肠易激综合征、肠结核、消化道肿瘤及其他恶性肿瘤者;(3)伴有严重的心、肝、肾功能不全,代谢性疾病以及其他严重疾病者;(4)近1个月内使用过抗生素,抑酸、胃动力药物、微生态调节剂、糖皮质激素及免疫抑制剂使用者;(5)最近1个月内发生过腹泻,行胃肠道手术或行胃肠镜检查者;(6)最近1个月内突然的生活环境和饮食结构的改变;(7)营养不良、免疫系统缺陷或先天性遗传代谢病;(8)有精神病史,服用镇静催眠药、滥用药物。两组患者一般资料比较差异无统计学意义(P>0.05),见表1。

表1 两组患者一般资料比较
1.2 方法
1.2.1 粪便样本获取 采集粪便样本前嘱患者排空尿液,无菌粪便收集器收集患者新鲜粪便,而后立即放入-80℃冰箱中保存备用。
1.2.2 粪便DNA提取及实时荧光定量PCR检测 采用16S rRNA测序方法检测患者的肠道菌群特征,使用E.Z.N.A.®Stool DNA试剂盒(生产厂家:美国Omega公司;货号: D4015-01)提取微生物DNA,并保存在-20℃,使用以下聚合酶链式反应(PCR)引物扩增细菌16S核糖体RNA基因的V3-V4区: 5′-ACTCCTACGGGAGGCAGCAG-3′(正向)和5′-GGACTACHVGGGTWTCTAAT-3′(反向)。PCR扩增使用TransStart®FastPfu聚合酶系统,反应混合物(包括5 × FastPfu Buffer 4 μL,dNTPs(2.5 mmol/L)2 μL,每个引物(5 μmol/L) 0.8 μL, FastPfu聚合酶0.4 μL,模板DNA 10 ng)在95℃初始变性2 min,然后进行25个循环,分别为95℃ 30 s, 55℃ 30 s,72℃ 30 s,最终延伸时间为72℃ 10 min。使用Silva 参考数据库预先训练的朴素贝叶斯算法分类器对菌群物种进行注释,去掉注释为线粒体、古生物、真核生物的序列,在QIIME2平台计算样本α多样性:包括菌群丰度指标(包括Chao1值和ACE值,其值越大表示物种总数越多)和菌群多样性(包括Shannon值和Simpson指数,Shannon值越大、Simpson指数越小群落表示菌群多样性越高);计算利用样本间基于 Bray-Curtis 距离的 β 多样性指标。
1.3 血液样本采集及代谢谱分析
1.3.1 血液样本采集 所有对象采集隔夜空腹外周静脉血5 mL,置于EDTA抗凝管中,室温静置1 h后离心(3 000 r/min,离心15 min),静置后取上层血清立即放入-80℃冰箱中保存备用。
1.3.2 GC-MS色谱分析 取同一受检溶液连续进样6次,进行色谱柱检测,色谱峰个数和相对峰面积RSD<5%,提示仪器精密度良好。采用脉冲式进样,温度为290℃,以氦气为载体,流速为1 mL/min,进样量为1 μL,升温程度设置为初始温度45℃,维持2 min,然后以9℃/min的梯度升高到180℃,再以40℃/min的梯度升高到240℃,维持12 min,在1 min内升高到280℃,维持2 min。每次分析前均需要等待6 min平衡色谱柱。采用超高效液相色谱-飞行时间质谱联用技术检测血清代谢小分子,将原始数据文件导入到Progenesis QI(Version 2.0, Waters, Milford, MA, USA)软件;经 Progenesis QI预处理的数据矩阵可直接导入具有扩展统计功能的EZInfo3.0软件(Waters, Milford, MA, USA)进行多元统计分析,从而筛选密切相关的血清差异表达代谢物。
1.4 代谢通路分析 (1)肠道菌群代谢通路分析:分析前先将代谢物浓度进行标准化处理后,将处理后的数据导入SIMCA-P 14.1 软件进行组间代谢物差异分析,得出有差异的代谢物后基于KEGG数据库,通过R语言包进行通路分析。(2)血清代谢通路分析:将在正负离子模式下鉴定的血清差异代谢物导入Metabo Analyst软件进行代谢通路分析。
1.5 统计学方法 计量资料以均数±标准差表示,组间两两比较采用t检验。基于分类学信息和多变量统计方法可对两组样本的菌群结构组成及物种差异等进行分析,采用SPSS 19.0软件与R软件进行统计分析。P<0.05为差异有统计学意义。
2 结果
2.1 两组肠道菌群多样性比较 肠道菌群α多样性方面,DM组Chao指数、Ace指数、Shannon指数低于对照组,Simpson指数高于对照组,差异有统计学意义(P<0.05);两组肠道菌群β多样性差异有统计学意义(P<0.05),见表2、图1。

图1 两组肠道菌群β多样性比较
表2 两组肠道菌群α多样性比较
2.2 两组肠道菌群结构比较 16S rRNA高通量测序后经过LEfSe分析显示,DM组患者肠道菌群中以肠球菌属(Enterococcus)、梭菌属(Clostridioides)、粪杆菌属(Faecalibacterium)、放线菌属(Actinomyces)、埃希菌属(Escherichia)、伊格尔兹菌属(Eggerthella)、安德克菌属(Adlercreutzia)占主要优势;对照组肠道菌群中以拟杆菌门(Bacteroidetes)、乳杆菌属(Lactobacillus)、双歧杆菌属(Bifidobacterium)、副杆菌属(Parabacteroides)、普雷沃菌(Prevotella)、甲烷短杆菌属(Methanobrevibacter)、二十碳孢梭菌属(Erysipelatoclostridium)占主要优势,见图2。

图2 两组患者粪便菌群LDA值分布柱状图
2.3 两组血清代谢标志物比较 基于偏最小二乘法判别分析模型(PLS-DA)模型,鉴别了10个代谢物,进行了相对浓度单维上的变化情况分析,结果显示:DM组乳酸、亚油酸、棕榈酸、油酸水平高于对照组,L-丙氨酸、L-缬氨酸、络氨酸水平低于对照组,差异有统计学意义(P<0.05),见表3。
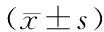
表3 两组血清代谢标志物水平比较单维相对浓度
2.4 肠道菌群和代谢标志物的代谢通路分析比较 PICRUSt工具对肠道差异菌群进行基因功能通路预测显示,差异菌属可获得三个代谢通路信息,分别是其他次级代谢产物的合成、脂质代谢、碳水化合物代谢;分析血清差异代谢物涉及的代谢通路显示,共有9条差异通路,其中涉及脂质代谢的通路最多;比较两种代谢通路,脂肪酸的生物合成通路为肠道菌群和血清代谢物的共有代谢通路。见图3。

图3 肠道菌群和代谢标志物的代谢通路分析比较
3 讨论
近年来随着人们对肠道菌群的认识不断加深,肠道菌群已经不再被认为是一个“闲置器官”,其复杂的微生态系统对于调节机体能量代谢、免疫功能及肠道黏膜完整性具有重要作用[7]。研究发现[8-9],肠道菌群通过参与胆汁酸代谢、胆碱代谢及生物拮抗作用参与机体糖脂代谢、细胞代谢及抵抗外来微生物定植和增殖。大量研究表明[10-11],肠道菌群功能失调与许多自身免疫性疾病、代谢性疾病、肿瘤及糖尿病并发症的发生有关。DM作为影响国人身体健康和生命安全的全身综合代谢性疾病,深入了解DM患者的肠道菌群特征,能够为DM的预防和干预提供新的治疗靶点。
研究显示[12],T2DM患者肠道菌群浓度更高,且结构及功能也与血糖正常人群不同,DM前期患者在菌属水平上,梭菌丰度显著下降,而萨特菌属及链球菌属丰度显著增加。本研究结果显示,DM组肠道菌群丰度和菌群多样性均优于对照组,且DM组以肠球菌、梭球菌属为主,对照组以拟杆菌属、乳杆菌属和双歧杆菌属为主,与目前研究相一致。分析认为:肠道菌群原有的生态平衡一旦被打破,机体的能量代谢、免疫功能及抵抗外来微生物入侵的能力将会受到影响。拟杆菌属、双歧杆菌属在肠道内能够产生一种名为丁酸的成分,可以为结肠上皮细胞提供能量,提高肠上皮细胞的完整性[13],且拟杆菌水平于体内内毒素水平呈负相关,拟杆菌水平较低的患者机体炎症反应程度越重[14]。当DM患者拟杆菌水平下降,肠道完整性丢失,炎症反应加重,而目前研究认为慢性炎症与DM发生相关[15]。短链脂肪酸作为肠道菌群特别是乳酸杆菌的主要代谢产物,被证实具有调节固有免疫和维持肠道黏膜屏障完整性的作用。短链脂肪酸能够通过抑制IL-12的产生,进而抑制促炎因子TNF-α和IL-1β引起的抗炎作用,在减轻DM导致的机体微炎症状态具有积极效果。此外,肠球菌水平与DM患者空腹血糖水平呈正相关[16],肠球菌数量的增加与DM血糖水平及DM并发症的发生存在重叠效应[17],但目前关于此方面的研究尚无明确结论。
在代谢组学方面,DM组与对照组在多项血清代谢指标方面存在差异,表明DM的糖脂代谢、氨基酸代谢途径进发生了不同程度的紊乱。DM及其并发症作为代谢组学研究中最重要的疾病之一,近年来有学者对DM的代谢谱做了进一步分析发现,T2DM患者在脂肪酸类代谢谱和血浆磷脂类代谢谱中与正常人群存在显著差异[18]。在本研究中我们对DM存在差异的代谢指标进行了代谢通路分析并与肠道菌群的代谢通路进行对比后发现,脂肪酸的生物合成通路为肠道菌群和血清代谢物的共有代谢通路。人体的代谢主要是由机体自身基因组和微生物基因组调节的代谢共同组成,肠道菌群中定植的细菌在分解宿主无法分解的物质同时还可参与宿主自身的物质代谢[19]。在DM患者中脂质代谢通路作为肠道菌群代谢和宿主自身代谢的共同通路均发生了不同程度的紊乱,笔者猜想肠道菌群可能通过影响DM患者脂质代谢来干预DM的发生发展。一项研究指出[20],肠道菌群中的某些微生物可以通过其代谢产物来介导某些神经递质的产生,还可通过调节胃肠激素如血清素、饥饿素、胰高血糖素等的分泌,参与宿主神经系统和内分泌系统的调节。而近期一项研究[21]显示,红芸豆多糖可通过调节肠道菌群及脂质代谢减轻T2DM大鼠血糖及血脂,而肠道菌群参与机体脂质代谢,提示肠道菌群可能通过影响脂质代谢从而影响患者血糖。
综上所述,T2DM患者存在肠道菌群失调,肠球菌属、梭菌属、粪杆菌属含量增多,拟杆菌属、乳酸杆菌和双歧杆菌属含量减少;且T2DM患者和健康人群的代谢指标存在差异,肠道菌群可能通过影响脂质代谢来影响T2DM的发展。但本研究限于样本量较小,后期还需增大样本量进一步论证;另外,本研究对于代谢组学方面的研究还不够深入,肠道菌群影响T2DM的代谢机制还需进一步深入研究。
利益相关声明:本文所有作者声明无利益冲突。
作者贡献说明:研究设计和论文写作为阿孜古丽·买合买提,研究方案执行与实施为米日古丽·吾木哈斯木、李雪梅和塞娜瓦尔·阿西木,数据统计分析及论文修改为穆叶斯尔·哈斯木,查文献和病例资料的整理为阿孜古丽·买合买提和穆叶斯尔·哈斯木。
