Identification of Fusarium wilt resistance gene SiRLK1 in Sesamum indicum L.
2024-03-07YinghuiDuanWenwenQuShuxianChangMingJuCuiyingWangCongMuHengchunCaoGuitingLiQiuzhenTianQinMaZhanyouZhangHaiyangZhangHongmeiMiao
Yinghui Duan, Wenwen Qu, Shuxian Chang, Ming Ju, Cuiying Wang, Cong Mu,Hengchun Cao, Guiting Li, Qiuzhen Tian, Qin Ma, Zhanyou Zhang, Haiyang Zhang,*,Hongmei Miao,*
a The Shennong Laboratory, Zhengzhou 450002, Henan, China
b Henan Sesame Research Center, Henan Academy of Agricultural Sciences, Zhengzhou 450002, Henan, China
c Key Laboratory of Specific Oilseed Crops Genomics of Henan Province, Henan Sesame Research Center, Henan Academy of Agricultural Sciences, Zhengzhou 450002, Henan, China
Keywords:Breeding for resistance Genetic analysis Molecular analysis Tandem kinase domain
ABSTRACT Sesame Fusarium wilt(SFW),caused by Fusarium oxysporum f.sp.sesami(Fos),is one of the most devastating diseases affecting sesame cultivation.Deciphering the genetic control of SFW resistance is pivotal for effective disease management in sesame.An inheritance study on a cross between the highly resistant variety Yuzhi 11 and the highly susceptible accession Sp1 using a Fos pathogenicity group 1 isolate indicated that resistance was conferred by a single dominant allele.The target locus was located in a 1.24 Mb interval on chromosome 3 using a combination of cross-population association mapping and bulked segregant analysis.Fine genetic mapping further narrowed the interval between 21,350 and 21,401 kb.The locus Sindi_0812400 was identified as the SFW resistance gene and officially designated SiRLK1.This gene encodes a specific malectin/receptor-like protein kinase with three putative tandem kinase domains and is considered a kinase fusion protein.Sequence analysis revealed that a high proportion(49.44%)of variants within the locus was located within the kinase domain III,and several of which were evidently associated with the diversity in SFW response,indicating the critical role of kinase domain III in expression of disease resistance.These findings provide valuable information for further functional analysis of SFW resistance genes and marker-assisted resistance breeding in sesame.
1.Introduction
Sesame(Sesamum indicum L.,2n=26)is an ancient nutritionally rich oilseed crop,with high contents of fatty acids (~55%), protein(~20%), and natural antioxidants including sesamin, sesamolin,and tocopherols.It is mainly cultivated in tropical and subtropical regions of Africa,Asia,and South and Central America[1].Over the past decade, there has been a consistent expansion of the sesame cultivation area, which made an increase in the total production.However, the yield of sesame remains low due to various biotic stresses, including more than 20 pests and diseases [2].Fusarium wilt (SFW), caused by the soil-borne fungus Fusarium oxysporum f.sp.sesami (Fos), is a particularly devastating threat [3].Widely present in traditional sesame-producing countries, SFW has an annual incidence of approximately 15%, leading to substantial losses in sesame seed yield [4–6].Controlling SFW using agricultural and chemical measures, such as crop rotation and fungicides is challenging as the Fos pathogen can remain viable in the soil,persisting as chlamydospores for more than six years.Consequently, the development and deployment of resistant varieties remain a preferred approach to manage SFW [7].
Over the past two decades, substantial effort has been made in evaluating the SFW response levels of sesame germplasms[2],and many accessions from different regions exhibited various degrees of resistance under natural infection and artificially inoculated conditions [8–11].However, the genetic basis of resistance is only partially explored.Both additive and non-additive effects have been implicated in previous inheritance studies [6,7,12].Resistance in several sesame cultivars was inferred to be controlled by 1 or 2 dominant genes[13].Despite these reports,the specific loci associated with response to SFW in sesame remain elusive.
In other species, such as Solanum pimpinellifolium, S.pennellii and S.lycopersicum,Cucumis melo,Arabidopsis thaliana,and Brassica oleracea,genes for resistance to Fusarium wilt have been identified using genetic mapping and high-throughput sequencing technology [14–20].These genes are dominant and confer resistance to specific formae speciales.Genes I, I-2, and I-3 in tomatoes encode a leucine-rich repeat receptor-like protein (LRR-RLP), a coiledcoil, nucleotide-binding, leucine-rich repeat protein (CC-NB-LRR),and an S-receptor-like kinase that confer resistance to F.oxysporum f.sp.lycopersici races 1, 2, and 3, respectively [15,17,20].
Three Fos pathogenicity groups were previously determined in the sesame-Fos pathosystem, based on markedly different pathogenicities to sesame host genotypes [5].Notably, varieties such as Yuzhi 11 and J9014 were immune or highly resistant to Fos pathogenicity group 1 isolates, whereas others such as Sp1 and HJ16 were highly susceptible.In this study, we revealed that resistance to Fos pathogenicity group 1 isolates was controlled by a major dominant gene,SiRLK1.This resistance gene was identified by fine mapping of genetic populations from a cross between Yuzhi 11 and Sp1.We found that SiRLK1 encodes a specific malectin/receptor-like protein kinase with three putative tandem kinase domains (TKDs), with the third kinase domain proving critical for expression of resistance.
2.Materials and methods
2.1.Plant materials and population construction
The SFW-resistant sesame variety Yuzhi 11, selected as a mutant of Yuzhi 4, and the highly susceptible accession Sp1 collected in Xinxiang by the Henan Sesame Research Center, Henan Academy of Agricultural Sciences were crossed to generate F1(reciprocal crosses), F2, BC1, and F6populations.A residual heterozygous line (RHL) was identified in the F6population though progeny testing,and self-pollinated to generate RHL-F7population segregating for response to SFW.The parents, F1, F2, and BC1populations were used for inheritance studies and separate F2and RHL-F7populations were used for genome sequencing and fine mapping of candidate resistance genes.Additionally, 142 sesame accessions,comprising 51 Chinese varieties and 91 accessions from China and 20 other countries,were randomly selected from sesame germplasm held by the Henan Sesame Research Center, for genespecific marker validation and haplotype analysis.
2.2.Pathogen inoculation and evaluation of SFW response
FS10175,a representative isolate of Fos pathogenic group 1,collected from Xuancheng was used to infect the host populations.This isolate was avirulent to some SFW-resistant accessions but highly pathogenic to others [5].Sesame seedlings at the four-leaf stage (4-week-old) were inoculated with FS10175 as described[10].Briefly,the roots of seedlings were dipped into a 5×106conidia mL-1microconidial suspension of FS10175 for 10 min and the plants were subsequently grown in growth chambers under a 12 h light/darkness cycle at 28/23 °C and 70% relative humidity).SFW symptoms were scored at 7, 14, 21, and 28 days post-inoculation(dpi), using a disease severity scale ranging from 0 to 4 (0 for immune or highly resistant, no visible symptoms; 1 for resistant,slight wilting of seedlings; 2 for moderately resistant, moderate wilting of leaves and slow growth of seedlings; 3 for susceptible,severe wilting or abscission of leaves and the noticeably slow growth of seedlings;and 4 for highly susceptible,complete wilting or abscission of leaves and death of the whole plant).For inheritance studies,more than 30 individuals of the parents and F1populations,and at least 90 F2and BC1plants were evaluated.For fine mapping of the candidate resistance gene, 15–30 individuals from each F3and RHL-F8line were evaluated to confirm the genotype of the corresponding F2and RHL-F7plant.In addition, to assess the responses of 142 sesame accessions,~30 individuals per accession were inoculated,and incidence and disease index(DI)of SFW were calculated as described [8,10].The response levels were graded according to the criteria:0 ≤DI ≤15,immune or highly resistant;15 < DI ≤30, resistant; 30 < DI ≤55, moderately resistant;55 < DI ≤70, susceptible; and 70 < DI ≤100, high susceptible.
2.3.Genomic DNA extraction and whole–genome sequencing
Genomic DNA was extracted from young leaves of the parents,F2, and RHL-F7populations using an improved cetyltrimethylammonium bromide method [21].Thirty-one homozygous resistant and 21 homozygous susceptible plants were chosen from the F2population (Sp1 × Yuzhi 11) for association mapping, and their genomic DNA was extracted for genome sequencing.DNA from 20 homozygous resistant and 20 homozygous susceptible plants from the RHL-F7population was separately pooled in equal amounts to form resistant and susceptible bulks for bulked segregant analysis (BSA).
DNA samples were sonicated to produce 350 bp-long fragments and standard paired-end libraries were constructed according to the Illumina TruSeq DNA sample preparation guidelines.Wholegenome resequencing of the libraries was performed using the Illumina NovaSeq 6000 platform by the Anoroad sequencing service(Anoroad Gene Technology, Beijing).The parents, each individual F2or RHL-F7plant, and bulks had sequencing depths of approximately 20-fold, 15-fold, and 100-fold, respectively.
2.4.Analysis of sequence data and localization of a candidate region
Raw reads from genomic sequencing were trimmed using Trimmomatic (v.0.33) and aligned to the Yuzhi 11 reference genome(v.3.0, PRJNA315784) using BWA (v.0.7.15) with default parameters [22].Single nucleotide polymorphism (SNP) and insertiondeletion (InDel) variants were detected using GATK joint calling analysis following the best-practice pipeline [23].Among them,variants from the above 52 individuals were filtered with the criteria of minimum variant count ≥ 31 and minimum allele frequency ≥0.05.The filtered variant call format file was used to conduct an association analysis of the variants with the SFW phenotypes in the F2population using the general linear model of TASSEL(v.5.2.43).A threshold of P value set as 1/total number of SNPs and InDels determined the candidate region(s)significantly associated with SFW resistance.
BSA was performed as previously described [24].SNP-index in the resistant and susceptible bulks and Δ(SNP-index) between the two bulks were calculated for all identified SNP and InDel positions, respectively.To reduce analysis errors, some variant positions were excluded according to the following criteria:heterozygous parent genotype, SNP-index value < 0.3 in both bulks, and sequencing depth < 10.SNP-index plots were drawn based on the SNP-index values and average SNP-index values calculated within a 100 kb increment and 1 Mb sliding window size.A 99%confidence level was set as the screening threshold to generate confidence intervals.
2.5.Fine mapping of the SFW resistance gene
Ten SNPs in the candidate regions were selected to develop high-resolution melting (HRM)–based SNP markers to fine-map the SFW resistance gene in the F2population.Primer pairs of the SNP markers were designed based on the Yuzhi 11 genome sequences of 200 bp before and after SNP physical positions using the Primer Premier 5.0 program (https://www.premierbiosoft.com/prierdesign/index.html).Details of the primer pairs are listed in Table S1.HRM analysis was conducted to genotype the F2mapping population and identify recombinants with the SNP markers on a LightCycler 480 Instrument II (Roche Diagnostics, Rotkreuz, Switzerland).DNA amplification was conducted using 20 μL of a mixture containing 1.0 μL of genomic DNA(20 ng μL-1), 10 μL of 2× High resolution melting master (Roche Diagnostics, Mannheim, Germany), and 0.2 μmol L-1of each primer.PCR amplification and gene scanning assays were conducted according to the instrument operator’s manual.
2.6.Cloning of the SFW resistance gene
SFW resistance genes were identified based on co-segregating SNP markers and Yuzhi 11 genome annotation.Genomic DNA extracted from young leaves of Yuzhi 11 and Sp1 was used to clone the DNA sequences of SiRLK1 and its allele, sirlk1.To obtain the cDNA sequences of SiRLK1 and sirlk1, total RNA was isolated from root tissues of Yuzhi 11 and Sp1 at 72 h post-inoculation with FS10175 using RNAiso Plus Reagent (TaKaRa, Dalian, Liaoning,China).The DNA was treated with DNase I to remove genomic DNA.cDNA was synthesized with oligo(dT)18primer using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific,Vilnius, Lithuania).To amplify the full coding sequences (CDS) of SiRLK1 and sirlk1,PCR and reverse transcription(RT)-PCR were performed on an Eppendorf Mastercycler (Eppendorf, Hamburg, Germany) using Phanta Max Super-Fidelity DNA Polymerase(Vazyme, Nanjing, Jiangsu, China) with the primer pair RLK1-F&R(Table S2),following the manufacturer’s instructions.The products were then subjected to Sanger sequencing on an ABI 3730 XL DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
2.7.Sequence analysis and phylogenetic tree construction
The sequence of SiRLK1 was queried using NCBI BLAST(https://blast.ncbi.nlm.nih.gov/Blast.cgi) to search for homologs and confirm its annotation.The putative protein sequence derived from the CDS of SiRLK1 was analyzed using Pfam (https://pfam.xfam.org/), SignalP (https://services.healthtech.dtu.dk/services/SignalP-4.1/), and Tmpred (https://www.ch.embnet.org/software/TMPRED_form.html)to predict conserved domains and motifs,signal peptides, and transmembrane helices.Fifteen homologs of the SiRLK1 protein in 12 other plants and I, I-3, and I-7 in tomatoes were found from the NCBI protein database (https://www.ncbi.nlm.nih.gov/protein/) and alignment of SiRLK1 with these homologs was performed using MUSCLE with default settings [25].The Neighbor-Joining tree was constructed using MEGA software(v.5.2,https://www.megasoftware.net/download_form)and tested using the bootstrap method with 1000 random replications.
2.8.Allelic variation
The 142 sesame accessions were genotyped with marker S3_21381019 to further validate the marker-trait association.Specific accessions were chosen for analysis of allelic variation of SiRLK1 based on marker genotypes that differed from Yuzhi 11 and Sp1, or different SFW responses.The SiRLK1 alleles were first assembled using genome resequencing datasets for these lines.Based on the alignment of assembles,representative sesame accessions were selected for further sequence verification by PCR.A universal PCR primer pair (listed in Table S2) was designed from the putative sequences to amplify all SiRLK1 alleles.PCR amplification,product sequencing, and sequence alignments were performed as described above.A neighbor-joining tree was constructed for SiRLK1 and its alleles in the sesame accessions.Selected accessions with non-repetitive sequences represented the different haplotypes.These accessions are shown in the schematic of sequence alignment and phylogenetic tree.
3.Results
3.1.Inheritance of SFW resistance in sesame
Phenotypic differences were evident between Yuzhi 11 and Sp1 post-inoculation with Fos isolate FS10175(Fig.1).The seedlings of Yuzhi 11 displayed immunity with an SFW incidence and DI of 0,whereas Sp1 exhibited high susceptibility with SFW incidence of 100% and DI of 100 from 14 dpi.To explore the genetic basis of SFW resistance in sesame,the segregating populations were generated using Yuzhi 11 and sp1.The inheritance studies shown in Table 1 indicated that resistance in Yuzhi 11 was conferred by a single dominant allele.Furthermore, the SWF responses of 109 F3and 83 RHL-F8lines were assessed, and the results revealed that the corresponding genotypic ratios of F2(31 homozygous resistant/57 heterozygous/21 homozygous susceptible, χ2= 2.064 <χ20.05,2) and RHL-F7(22 homozygous resistant/40 heterozygous/21 homozygous susceptible,χ2= 0.133 <χ20.05,2) fitted 1:2:1.
3.2.Location of a candidate chromosome region associated with SFW resistance
We conducted a combined analysis of cross-population association mapping(CPAM)of the F2population to locate the SFW resistance gene.Thirty-one homozygous resistant and 21 homozygous susceptible F2individuals and parents were sequenced using the Illumina platform.A total of 5.98 Gb and 9.61 Gb of clean data from Yuzhi 11 and Sp1 were generated with 18.72-fold and 30.19-fold genome coverage (Yuzhi 11 genome size of 311.23 Mb), and that from F2individuals ranged from 4.14 to 7.40 Gb with an average coverage of 18.55-fold(Table S3).All clean reads from the 54 samples were aligned with the sesame reference genome for SNP and InDel detection.A total of 402,719 SNPs and InDels with high confidence were screened by joint calling and original variant set filtering, and then plotted on the 13 sesame chromosomes(Fig.2A).CPAM identified a genomic region at 13.08 to 24.33 Mb in chromosome 3 (P ≤ 2.48 × 10-8) with 1208 variants(Table S4).Among these, 106 variants with the lowest P value(P = 0) were located in the region between 21.29 and 22.29 Mb(Table S4).
BSA confirmed the genomic region associated with SFW resistance using the resequencing dataset of the R and S bulks from the RHL-F7population (Table S3).A total of 5,586 high confidence SNPs and InDels were detected across the 13 chromosomes.SNPindex and Δ(SNP-index) (S bulk - R bulk) values calculated and plotted on the chromosomes identified a candidate gene region bordered by 20.89 and 22.52 Mb of chromosome 3 with 99% confidence level of the average Δ(SNP-index) (Fig.2B).The region overlapped with the partial candidate interval of CPAM (Fig.2;Table S5).The peak of the region was located in an interval between 21.10 and 22.34 Mb with a Δ(SNP-index) value of 1.0.Combining the results of CPAM and BSA, a genomic region of 1.24 Mb was finally determined to be linked to the SFW resistance gene locus,which contained 150 variants with P ≤1.87×10-9and Δ(SNP-index) = 1.
3.3.Identification of a candidate resistance gene by fine mapping
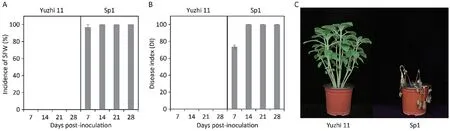
Fig.1.Phenotypic characteristics of the resistant variety Yuzhi 11 and susceptible accession Sp1 inoculated with the Fusarium oxysporum f.sp.sesami isolate FS10175.(A)Incidence and(B)disease index of sesame Fusarium wilt(SFW)calculated at 7,14,21,and 28 days post-inoculation(dpi).(C)SFW responses of Yuzhi 11 and Sp1 plants at 14 dpi.

Table 1 Phenotypic statistics and inheritance analysis of Fusarium wilt resistance in Yuzhi 11 and Sp1.
Ten HRM-based SNP markers developed in the candidate genomic region were genotyped in a population of 225 F2individuals(Fig.3).Twenty recombinants were identified in the F2individuals based on a comparison of genotypes inferred from SNP markers and phenotypes of the corresponding F3lines.Of these recombinants,the homozygous genotypes of plants F2-66 and F2-168 were inconsistent with genotypes of markers S3_21350331 and S3_21401286, respectively.Correspondingly, the genotypes of markers S3_21381019 and S3_21381440 co-segregated with the phenotypes of all tested F2individuals.These results delineated the candidate region between S3_21350331 and S3_21401286,locating the candidate gene to an interval of 50.96 kb (21,350–21,401 kb) on chromosome 3 (Fig.3).
Only three variants including two co-segregating SNPs(S3_21381019 and S3_21381440) and SNP S3_21381606 within the identified interval were significantly associated with SFW resistance (P = 0).Further annotation of the variants revealed three SNPs in gene Sindi_0812400 encompassing T to A, A to G, and A to T missense mutations(Fig.4A).Therefore, this gene was identified as the target gene controlling resistance to the Fos isolate FS10175.
3.4.Characterization of the resistance gene SiRLK1
To further characterize the SFW resistance gene,the entire DNA and cDNA sequences of Sindi_0812400 and its allele from Yuzhi 11 and Sp1 were amplified using PCR and RT-PCR, respectively.The gene sequence in Yuzhi 11 was 5949 bp and contained 11 exons,with a complete CDS of 4467 bp (Fig.4A).It encodes a protein of 893 amino acids including an N-terminal signal peptide, two malectin-like domains, a transmembrane helix (TM), and three TKDs.BLAST analysis revealed that it had homology with of the malectin/receptor-like protein kinase gene At5g39000 in A.thaliana, a member of the CrRLK1L subfamily [26].The candidate gene was therefore designated SiRLK1 (GenBank accession no.MN604397).Three SNPs (T4684A, A4850G, and A5271T) relative to resistance allele were detected in the seventh,eighth,and ninth exons of sirlk1 allele in Sp1 resulting in non-synonymous mutations (E1198V, R1226Q, and V1336D) in the sirlk1 protein(Fig.4).All three mutations occurred in kinase domain III.
Alignment of SiRLK1 with its homologs showed conservation of the typical CrRLK1L protein sequence containing a malectin-like domain,TM,and the kinase domain I in Yuzhi 11 and 12 other species, with identity ranging from 34.40% (FLR11 in Oryza sativa) to 75.27% (PfRLK in Paulownia fortunei) (partial results in Fig.S1).The kinase domain II of SiRLK1 had a high similarity to those of its homologs in asterids with highest identity of 59.04%.The three TKDs adjacent to the TM and malectin-like domains appeared to be unique to Lamiales plants, which were only found in SiRLK1 in sesame and SaMRLK in Striga asiatica.Of the TKDs in the SiRLK1,kinase domain III was highly similar to kinase domain II (identity of 57.82%) but had a lower similarity to kinase domain I (identity of 36.65%) (Fig.S2).
Phylogenetic analysis indicated that SiRLK1 and its nine homologs in asterids, along with the six members of the CrRLK1L subfamily, formed a cohesive cluster.Notably, SiRLK1 and its eight homologs in the Lamiales were distinctly grouped in a subclade within this larger arrangement(Fig.5).In contrast,there was considerable evolutionary divergence between SiRLK1 and the known Fusarium wilt resistance proteins I,I-3,and I-7 in tomatoes(Fig.5).
3.5.Allelic variation at the SiRLK1 locus
To investigate whether the variants of SiRLK1 can explain the variation in SFW resistance in natural populations, the cosegregating marker S3_21381019 was used to genotype 142 sesame accessions, in addition to Yuzhi 11 and Sp1 (Fig.6).The genotypes of 98 accessions were identical to those of Yuzhi 11 or Sp1,and the SFW responses of 96 accessions were consistent with their phenotypes.The remaining 44 accessions had genotypes different from those of Yuzhi 11 and Sp1 within the amplified sequence of marker S3_21381019.Among them, the genotypes of the 28 resistant accessions were similar, represented by a Fos differential host J9014 [5].Therefore, the marker S3_21381019 genotypes predicted the SFW responses of 87.32% (124 of 142) of accessions, where the genotypes of Yuzhi 11 and J9014 predicted resistance, and the genotype of Sp1 predicted susceptibility.

Fig.2.Combined cross-population association mapping(CPAM)and bulked segregant analysis(BSA)for locating the candidate region associated with sesame Fusarium wilt resistance.(A)Manhattan plot from CPAM with 402,719 filtered single nucleotide polymorphisms(SNPs)and insertion-deletions(InDels).The threshold(red line)was set as P=2.48×10-8.(B)Δ(SNP-index)plot on chromosome 3 from BSA.Δ(SNP-index)was calculated by subtracting SNP-index of R bulk from SNP-index of S bulk.Average SNPindex values with 100 kb increments and 1 Mb sliding window size are plotted as the blue line.Red lines represent the 99%confidence interval.Gray shadows indicate the overlapping candidate interval contained in the variants with P ≤1.87 × 10-9 and Δ(SNP-index) = 1.
The presence of multiple genotypes in sesame accessions prompted the hypothesis that there were additional variants beyond the three identified SNPs between SiRLK1 and sirlk1.To confirm the allelic variation, the alleles of SiRLK1 were amplified from 20 representative accessions.This group included J9014,TP114, two accessions (YM121 and YM768) whose phenotypes were inconsistent with their genotypes of the marker S3_21381019, and 16 other accessions with diverse genotypes compared to Yuzhi 11,Sp1, and J9014.Sequence alignments identified 269 SNPs and InDels in the entire SiRLK1 gene(from the start codon to the stop codon) among Yuzhi 11, sp1, and the 20 accessions, 178 of which existed in the CDS, and 127 resulted in nonsynonymous or stop-lost mutations (Figs.7, S3).Remarkably,19.10%(34 of 178)and 49.44%(88 of 178)of variants were located in kinase domains II and III, respectively (Table S6).Based on the sequence variations, SiRLK1 and its alleles were divided into 13 haplotypes(Figs.7,S3).Further phylogenetic analysis placed these haplotypes into three clades (Fig.S4).The clade including haplotypes of Yuzhi 11, Sp1, J9014, YM121, TP114, TP130, and TP012 accounted for 126 of the 144 accessions.In addition to the three SNPs between SiRLK1 and sirlk1, several other SNPs in the kinase domain III were associated with the variations in SFW responses of sesame accessions (Fig.7).For instance, two non-synonymous nucleotide substitutions (A4684T and C5532G) between the resistant haplotype_YM121 and the susceptible haplotype_sp1 were critical for SFW resistance.In contrast, most variants located in the SP, malectin-like, and kinase I and II domains were detected in the resistant accession TP001(Fig.S3),suggesting that they were not related for SFW resistance.
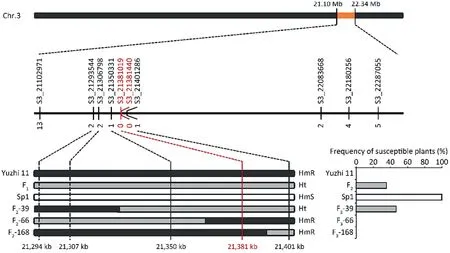
Fig.3.Fine genetic mapping of the candidate sesame Fusarium wilt (SFW) resistance gene using of 225 F2 individuals.Numbers below the bar indicate the numbers of recombinant plants between genotypes inferred from the markers and SFW responses of the corresponding F3 lines.The candidate gene was located within a 50.96 kb genomic region between the markers S3_21350331 and S3_21401286.Red dashed line indicates co-segregating markers S3_21381019 and S3_21381440.Black,gray,and white bars represent the homozygous resistant, heterozygous, and homozygous susceptible genotypes, respectively.

Fig.4.Sequence structure of the SiRLK1 and sirlk1 alleles.(A)Schematics of the structures of SiRLK1 in Yuzhi 11 and sirlk1 in Sp1.Solid bars and black lines indicate exons and introns, respectively.The bars in other than gray color represent the indicated domains.(B) Sequence alignment of kinase domain III in SiRLK1 and sirlk1.
4.Discussion
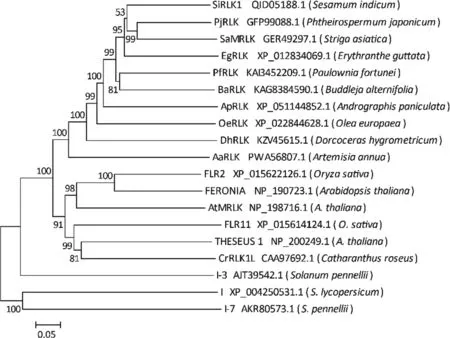
Fig.5.Phylogenetic analysis of SiRLK1 in sesame with 15 homologs and three Fusarium wilt resistance proteins in 14 other plants.The phylogenetic tree was constructed using the Neighbor-Joining method.Numbers at the nodes represent bootstrap values with 1000 random replications.Scale bar below the tree indicates the number of nucleotide substitutions.

Fig.6.Genotyping of marker S3_21381019 in a germplasm panel comprising Yuzhi 11,Sp1,and 142 other sesame accessions.The blue,green,and orange bars represent the genotypes of Yuzhi 11,Sp1,and J9014, respectively; the yellow bars represent other genotypes.Yuzhi 11 and J9014 have been selected as the differential hosts to Fusarium oxysporum f.sp.sesami(Fos)[5];Yuzhi 11 is resistant to Fos pathogenicity group 1 and 2,while J9014 is highly resistant to Fos pathogenicity group 1 but highly susceptible to Fos pathogenicity group 2.The vertical coordinate value of each bar indicates the disease index of the accession.The resistance levels of sesame accessions were evaluated according to the criteria as described [10].
Resistance to SFW has become the primary objective in sesame breeding.The identification of SFW-resistant germplasms and genetic analysis of resistance are crucial in addressing this challenge,as currently,the genetic basis of resistance to SFW in sesame is poorly understood.Across plant species Fusarium wilt resistance is conferred by dominant genes [18,20,27] and quantitative trait loci (QTL) [28,29].Resistance to SFW in sesame was previously hypothesized to exhibit the quantitative responses with both additive and non-additive gene actions [6,12].The apparent contradiction in findings across different experiments has been attributed to different genetic materials, pathogenic strains, inoculation methods, and environmental influences.We elucidated that the genetic pattern of SFW resistance in Yuzhi 11 was controlled by a single dominant gene through classical genetic analysis of the cross-population.A prerequisite for this analysis involved the use of a single isolate, FS10175, from Fos pathogenicity group 1, along with a dependable SFW response evaluation method [5,10].This finding was consistent with our prior hypothesis of gene-forgene resistance in sesame [5].Moreover, the clear phenotypic differences among most sesame accessions, characterized by either high resistance or susceptibility to the Fos isolate clearly deviated from a continuous distribution (Fig.6).This observation is compelling evidence supporting a qualitative gene mode.

Fig.7.Allelic variations in the SiRLK1 locus.The sequence of SiRLK1 was used as the reference.Yuzhi 11, Sp1, and other 11 diverse sesame accessions were selected to represent the 13 different haplotypes of SiRLK1.Variants in the kinase domain III are shown below the structural schematic.
We identified the SFW resistance gene SiRLK1 using a combination of genome resequencing and genetic mapping.To accurately identify the candidate region associated with SFW resistance,CPAM and BSA were applied to the F2and RHL-F7populations,respectively.The combined approach delimited the locus to a 1.24 Mb genomic region.BSA was considered the preferred method of mapping because of its advantages in terms of convenience,cost-effectiveness, and efficiency.
SiRLK1 is the first resistance gene cloned in sesame and is different from the NB-LRR and protein kinase Fusarium wilt resistance genes identified in other plant species [14–20].SiRLK1 in sesame variety Yuzhi 11 encodes a malectin/receptor-like protein kinase,considered to be a member of the CrRLK1L subfamily based on sequence alignment and phylogenetic analysis.CrRLK1L proteins have been shown to play versatile roles in plant immunity and stress response, as well as growth, development, reproduction,and hormone signaling [30].Arabidopsis FERONIA (FER), the best-characterized CrRLK1L member, acts as a rapid alkalinization factor(RALF)-regulated scaffold,orchestrating the intricate assembly of immune receptors EFR and FLS2 with the co-receptor BAK1 to suppress plant immune responses [31].Correspondingly, the absence of FER in Arabidopsis plants enhances resistance to F.oxysporum[32].In addition to FER,two other FER-like receptors in rice,FLR2 and FLR11, were found to be negative immune receptors for resistance against rice blast[33].In contrast,THESEUS 1 positively regulates defense response to Botrytis cinerea via the downstream GEF4 signaling pathway [34].SiRLK1 is also a positive immune modulator in the CrRLK1L subfamily, as it exhibits complete dominance in conferring resistance to SFW.Nevertheless,the relatively large phylogenetic distance between SiRLK1 and CrRLK1L members suggests functional differences.
SiRLK1 exhibits high sequence conservation with its homologs in the Lamiales.Specifically, SiRLK1 manifests as a kinase fusion protein,encompassing the conventional CrRLK1L protein architecture with two malectin-like domains, a transmembrane domain,and a kinase domain.SiRLK1 diverges by including two additional TKDs, a distinctive feature unique to Lamiales.The discovery of TKD-containing proteins as a new family of disease resistance proteins in Triticeae species is noteworthy, with most members conferring resistance to fungal diseases [35,36].Several resistance genes in this family, such as Rpg1, Un8, WTK1, WTK2, WTK3, and WTK6-vWA, encode tandem protein kinases fused to different domains[35–40].Further phylogenetic analyses have revealed that TKDs originated from kinase domain fusions or kinase domain duplications [35].The kinase domains II and III of SiRLK1 exhibit a relatively high sequence identity(57.82%)to each other but have lower identity (36.65%) with kinase domain I.In addition, three SiRLK1 paralogs were identified among sesame-encoded proteins;each with a single fusion kinase domain, followed by the conventional CrRLK1L protein (data not shown).This suggests that kinase domains II and III may have originated from a duplication,whereas kinase domain I possibly had a different origin.Therefore,SiRLK1 is assumed to be derived from the fusion of an ancient CrRLK1L homolog and a kinase domain with a differing origin,coupled with a duplicated second kinase domain.
A comprehensive analysis revealed the presence of 178 SNPs and InDels within the CDS of SiRLK1 and its haplotypes in sesame accessions exhibiting different resistance levels; 68.54% of these variants were within the kinase domains II and III, implying that these domains were subjected to selective pressure during the evolutionary trajectory of SiRLK1.Additionally, three nonsynonymous mutations (T4684A, A4850G, and A5271T) identified in kinase domain III were associated with loss of resistance to SFW relative to the resistant variety Yuzhi 11.Conversely, two nucleotide substitutions (A4684T and C5532G) within the same domain conferred resistance relative to the susceptible accession Sp1.These findings point to a pivotal role of kinase domain III in SiRLK1, not unlike the significance of TKDs in other characterized kinase fusion proteins.Such TKDs play important roles in regulating signal transduction related to disease resistance [34,36,41].However, the specific function of SiRLK1 remains as a hypothesis based on its gene characteristics, which needs to be experimentally confirmed.Further functional analyses should be also conducted to elucidate the underlying molecular mechanism of SFW resistance.
In conclusion, we identified the first dominant SFW resistance gene in sesame, designated SiRLK1.This gene encodes a specific malectin/receptor-like protein kinase with multiple TKDs.Our research revealed a correlation between variation in kinase domain III and diversity in SFW response.These findings provide a foundation for comprehensive functional analyses of SFW resistance genes in sesame.Moreover, a marker S3_21381019 developed based on the variation within the SiRLK1 locus will facilitate marker-assisted breeding for the improvement of sesame disease resistance.
CRediT authorship contribution statement
Yinghui Duan:Conceptualization, Methodology, Writing –original draft.Wenwen Qu:Investigation, Validation.Shuxian Chang:Investigation.Ming Ju:Investigation.Cuiying Wang:Visualization.Cong Mu:Investigation.Hengchun Cao:Data curation.Guiting Li:Software.Qiuzhen Tian:Investigation.Qin Ma:Resources.Zhanyou Zhang:Resources.Haiyang Zhang:Supervision, Funding acquisition.Hongmei Miao:Writing – review &editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by China Agriculture Research System(CARS-14), the Key Research and Development Project of Henan Province(221111520400),the Henan Provincial Science and Technology Research Project (222102110081), the Zhongyuan Science and Technology Innovation Leading Talent Plan (214200510020),the Key Research Project of the Shennong Laboratory (SN01-2022-04), and the Fund for Distinguished Young Scholars from Henan Academy of Agricultural Sciences (2022JQ01).
Appendix A.Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2023.12.004.
杂志排行

The Crop Journal的其它文章
- Flowering-time regulation by the circadian clock: From Arabidopsis to crops
- Global characterization of OsPIP aquaporins reveals that the H2O2 transporter OsPIP2;6 increases resistance to rice blast
- Drought-triggered repression of miR166 promotes drought tolerance in soybean
- The OsBSK1-2-MAPK module regulates blast resistance in rice
- Natural variation of an autophagy-family gene among rice subspecies affects grain size and weight
- Rice gene OsUGT75A regulates seedling emergence under deep-sowing conditions