Identification and fine mapping of qSW2 for leaf slow wilting in soybean
2024-03-07ShengyouLiChanglingWangChunjuanYanXugangSunLijunZhangYongqiangCaoWenbinWangShuhongSong
Shengyou Li, Changling Wang, Chunjuan Yan, Xugang Sun, Lijun Zhang, Yongqiang Cao*,Wenbin Wang*, Shuhong Song
Institute of Crop Research, Liaoning Academy of Agricultural Sciences, Shenyang 110161, Liaoning, China
Keywords:Drought GWAS Linkage mapping Slow wilting Soybean (Glycine max)
ABSTRACT Drought is one of the abiotic stresses limiting the production of soybean(Glycine max).Elucidation of the genetic and molecular basis of the slow-wilting (SW) trait of this crop offers the prospect of its genetic improvement.A panel of 188 accessions and a set of recombinant inbred lines produced from a cross between cultivars Liaodou 14 and Liaodou 21 were used to identify quantitative-trait loci (QTL) associated with SW.Plants were genotyped by Specific-locus amplified fragment sequencing and seedling leaf wilting was assessed under three water-stress treatments.A genome-wide association study identified 26 SW-associated single-nucleotide polymorphisms (SNPs), including three located in a 248-kb linkage-disequilibrium (LD) block on chromosome 2.Linkage mapping revealed a major-effect QTL,qSW2, associated with all three treatments and adjacent to the LD block.Fine mapping in a BC2F3 population derived from a backcross between Liaodou 21 and R26 confined qSW2 to a 60-kb interval.Gene expression and sequence variation analysis identified the gene Glyma.02 g218100, encoding an auxin transcription factor, as a candidate gene for qSW2.Our results will contribute significantly to improving drought-resistant soybean cultivars by providing genetic information and resources.
1.Introduction
Soybean (Glycine max) provides protein and oil for human and animal consumption [1].Soybean production is impaired by drought, particularly in arid and semi-arid regions [2].Genetic improvement offers a key to increasing drought tolerance.This approach is particularly valuable in regions with high irrigation costs, limited access to irrigation water, or a lack of irrigation infrastructure.
Slow canopy wilting in soybean is a promising trait for improving drought tolerance.Soybean plants experience canopy wilting in response to drought stress,losing turgor and dropping their leaves.Soybean germplasm shows variation in wilting-onset time and severity [3,4].The slow-wilting (SW) trait is characterized by a delayed wilting response to a decrease in soil moisture content,relative to the common soybean cultivar.SW conserves soil moisture, which is more rapidly exhausted by fast wilting (FW) genotypes [5,6].Simulation models predicted [7] that the SW trait would increase yield under drought stress by more than 80% in most soybean-growing regions of the USA.Cultivars with the SW trait have been used as sources of SW in breeding programs [8].New cultivars show less wilting than older ones [9,10].
Identification of quantitative-trait loci (QTL) can help plant breeders develop superior cultivars.QTL analysis for SW have yielded a valuable approach for the identification of prospective genomic areas, as well as candidate genes or QTL associated with characteristics of significance.Four QTL for SW were mapped in a population of recombinant inbred lines (RILs) developed from a cross between KS4895 and Jackson.These QTL were located on chromosomes 8, 13, 14, and 17, and together accounted for 16%–47%of observed phenotypic variance in canopy wilting[11].Seven QTL were detected in five environments in 150 RILs developed from a cross of Benning × PI 416937 and accounted for about 75%of observed variation in canopy wilting[12].In five biparental mapping populations,eight QTL clusters containing QTL for canopy wilting were identified in at least two populations [13].Ye et al.[14] found two major QTL for SW located on chromosomes 6 and 10 in a cross of Magellan × PI 567731, and proposed a method for cloning them using near-isogenic lines.Soybean canopy wilting-based genome-wide association studies (GWAS) identified 61 SNPs in a panel of 373 accessions[9],45 in 162 accessions[10],and 188 in 200 accessions [16].These studies identified similar genomic regions on chromosomes 1, 4, 6, 9, 12, 15, 18, 19, and 20.Loci on chromosome 2 coincided with a previously reported[15] meta-QTL.
The objective of this study was to identify SNPs associated with SW in a GWAS, identify QTL associated with SW by QTL linkage mapping, fine-map a major-effect QTL using a BC2F3population,and identify a candidate gene for the QTL.
2.Materials and methods
2.1.Plant materials and drought-stress treatments
The 188 accessions of a soybean germplasm panel for GWAS(Table S1) consisted of 95 genotypes sourced from the Northeast soybean ecological zone in China, 48 genotypes from the Huang-Huai-Hai region in China, and 45 genotypes from various other countries.Of the 188 accessions, 49 were landraces and 139 improved cultivars.Two of these, Liaodou 14 (L14) and Liaodou 21(L21)were crossed to develop 178 F6:7RILs for linkage mapping.
Drought conditions were induced using three treatments: 10%and 20% PEG6000(10%PEG and 20%PEG), and water stress (WS).Treatments were imposed in an open field with auto-rain-shelter conditions at the Liaoning Academy of Agricultural Sciences in Shenyang(123°33′46′′E,41°49′54′′N),Liaoning province,China,during the 2019 cropping season.The automated rain shelter was opened in sunny weather and closed during rain.Phenotypic evaluations of the association panel and RILs were conducted in July and August,respectively.In both experiments, seeds were planted in a germination box(19×13×12 cm)containing 1.5 kg of quartz sand supplemented with 150 mL of 1/2 Hoagland nutrient solution.The seeds were allowed to germinate until the first trifoliolate leaf emerged.Drought stress was then imposed by addition of 100 mL of PEG6000solution with concentrations of 10% and 20%.In the WS treatment,the box contained 1.5 kg of soil matrix,maintained at an 80% moisture level until the first trifoliolate leaf emerged.Irrigation was then halted until leaf wilting was seen in 70% plants.For consistency, the seedlings were thinned to 10 per box in each treatment group.Each treatment was replicated three times for each accession and RIL, resulting in a minimum of 30 plants per treatment group.
2.2.Descriptive statistics and phenotypic evaluations
Three days after PEG treatments and five days after cessation of irrigation,leaf wilting scores under 10%PEG,20%PEG and WS conditions were recorded on a scale of 0–100,where 0 represented no wilting, 25 cotyledonary wilting, 50 both unifoliolate and cotyledonary wilting, 75 wilting of the first trifoliolate leaf, unifoliolate,and cotyledon,and 100 wilting of the whole plant.The leaf wilting index was calculated as follows:Σ(score×number of plants with each score)/total number of plants.
Correlations between wilting indices among treatments were calculated with SPSS 19.0 (SPSS, Inc., Chicago, IL, USA), and the variances were examined using the AOV function in QTL IciMapping (version 4.2) [17].The broad sense heritability (H2) of each variable was computed based on the ANOVA table [17].
2.3.Genotyping of soybean germplasm and RILs
DNA extraction was performed on leaves harvested around 60 d following germination, using a modified CTAB procedure [18].Specific-locus amplified fragment sequencing (SLAF-seq) [19],developed by Biomarker Technologies in Beijing, China, was used to create and analyze SLAF libraries from the 188 soybean accessions and 178 RILs.Restriction enzymes RsaI and HaeIII (NEB, Ipswich, MA, USA) were used.To distinguish raw sequencing data from digested fragments, adenine was added to the 3′end of digested fragments [20].SLAF tags were created by digestion of each soybean accession, fragment ligation, PCR amplification, and selection of target fragments for constructing SLAF libraries.After quality certification, including sequence quantity, base distribution,and sequencing quality distribution,SLAF-seq was performed on an Illumina HiSeqTM 2500 instrument(Illumina,Inc.,San Diego,CA,USA).The accuracy of SLAF libraries were assessed by comparative analysis with rice libraries (Oryza sativa L.ssp.japonica cv.Nipponbare) libraries (https://rice.plantbiology.msu.edu/), which were produced and sequenced using identical protocols.
We followed a standard protocol for grouping and genotyping the SLAF-seq data in order to ensure the quality of the bioinformatics analysis We compared the filtered sequencing reads with the reference genome using the BWA software (https://bio-bwa.sourceforge.net/) [21].To categorize SLAF producers into polymorphic,non-polymorphic, and repetitive groups, the analysis included allele frequencies and gene sequence variations.SLAF tags were used to identify polymorphic SNP loci mostly using GATK [22].SNP detection was also performed with SAMtools [23], to validate the accuracy and dependability of the SNPs found by GATK.SNPs with minor-allele frequencies (MAF) > 0.05 and marker integrity frequencies > 80% were chosen [24].
2.4.Genome-wide association studies
Population structure was analyzed using 67,929 SNPs in the 188-member panel.The ADMIXTURE software [25] was used to identify the hypothetical number of subgroups (K values).The tested K range of populations was 1 to 10 via the examination of population structure, which was repeated 1000 times.Each cross-validation (CV) error was calculated.Using the valley value of CV error rates, the optimal number of subgroups was determined according to cluster results [26].The analysis included the examination of the phylogenetic connections among 188 accessions, using the 67,929 SNPs.A distance matrix was used to compute the genetic distances between the materials using SNP markers.A phylogenetic tree was created with Tree Best [27](v1.9.2) by the neighbor-joining method.Linkage disequilibrium(LD) was calculated based on marker pairs located within whole genome by using PopLDdecay software [28].LD decay was measured as the chromosomal distance at which the average pairwise correlation coefficient(r2)decreased to half of its maximum value.
TASSEL software ver.5.0 [29] was used to fit a general linear model (GLM) to test for association between each SNP and trait.The mathematical representation of GLM is given by the equation y=Xb+e.In the present scenario,y represents the wilting index,X denotes the known design matrix,b represents a fixed-effects vector, and e is a vector of residuals.The threshold for the P-value,adjusted using the Bonferroni correction, was calculated as 0.1 divided by the total number (67,929) of tests conducted.This adjustment was made to account for multiple comparisons, with α representing the significance level set at 0.1.To ensure simplicity, a cutoff value of P < 1.47E-06 was employed.To reduce false positives,a GWAS was performed using a compressed mixed linear model (CMLM) that incorporates both population structure and kinship.For the CMLM, population structure and kinship were as a fixed effect and random effect, respectively.
2.5.Linkage mapping and QTL analysis
In the process of constructing linkage maps,a total of 6167 SNPs were selected, with all 20 chromosomes that exhibited more than 10%missing data or segregation distortion being excluded from the analysis (Fig.S2).The numbers of SNPs varied by chromosome,with chromosome 11 carrying 112 SNPs and chromosome 17,680 SNPs.The cumulative length of the linkage maps was 3766 cM,with a mean distance between markers of 0.61 cM.
To construct a genetic map, an input file was generated with QTL IciMapping [30] after redundant markers were removed.To order high-density markers, a combination of nearest neighbor and 2-Opt algorithms[31]was used.In an individual environmental analysis,the BIP function[32]was used with inclusive composite interval mapping(ICIM)to detect additive QTL.A LOD threshold of 2.5 was employed,and a 1000-permutation test was conducted at a 95%confidence level.The ICIM approach in the MET functional module [33] was used to identify QTL additive and QTL × environment interaction effects.QTL were named based on the truncated English name of the trait, preceded by the letter q and followed by the chromosome number.Where multiple QTL shared a chromosome, an order number was added.
2.6.Fine-mapping in a BC2F3 population
Based on the results of QTL analysis, one RIL, R26, was selected for backcrossing with the parent L21 to generate a BC2F3population for QTL fine-mapping.R26 shared 54% of its genetic background with L21 but carried the L14 allele associated with lower wilting index at a major-effect QTL for SW.Seeds of the BC2F3population and parents were grown in plastic pots of 12×12×12 cm filled with soil.DNA was isolated from 3526 plants of the BC2F3population.To genotype each plant, informative Kompetitive Allele-Specific PCR (KASP) markers [34] were developed based on SNPs identified by deep resequencing of L14 and L21 (Table S2).KASP assays were performed in 1536-well plates (LGC Genomics(LGC, Middlesex, UK).Fluorescence signals were measured with a Synergy H1 full-function microplate reader (FLUOstar Omega,BMG Labtech, Germany).Following the identification of seven key informative recombinants, the seedlings were transferred into pots of 30 × 30 × 25 cm containing 15.0 kg of soil, for phenotypic assessment.WS treatment was imposed 40 d after germination and was sustained for 6 d, with soil moisture content maintained at 50% of field water-holding capacity.Five leaves were sampled from each plant to measure relative water content (RWC) as RWC (%) = (FW-DW)/(TW – DW) × 100%, where FW, TW, and DW denote respectively fresh weight, turgid weight after immersion in water for 3 h, and dry weight.
2.7.qRT-PCR assessment
For measuring candidate-gene expression,24-h post-treatment leaves were sampled at the V3(three unfolded leaflets)stage from L14, L21 and R26 seedlings grown under 20% PEG and wellwatered (control) conditions in a greenhouse.A PrimeScriptRT reagent kit with gDNA Eraser (TaKaRa Bio Inc., Otsu, Japan) was used to synthesize cDNA from leaves.To conduct quantitative real-time PCR (qRT-PCR), a Light Cycler 480 II (Vazyme Biotech Co., Ltd., Nanjing, Jiangsu, China) was used.The primers used to amplify three candidate genes and the actin gene are described in Table S3.Three biological and four technical replications were used to calculate cycle threshold (CT) values.Fold changes of candidate genes between control and 20% PEG treatments were calculated by 2-ΔΔCT [35].
3.Results
3.1.Leaf wilting indices in association panel and RIL population
The frequency distribution of leaf wilting index in the 188-accession panel under the three water-stress treatments is shown in Fig.1A.The two parents L14 and L21 showed differing leaf wilting indices.Those of L14 were respectively 3.33, 6.67, and 3.33 under the 10%PEG, 20%PEG and WS treatments, and those of L21 were 96.67, 93.33 and 93.33.The frequency distribution of leaf wilting index in the 178 RILs derived from these parents is shown in Fig.1B.For both association panel and RILs, the leaf wilting indices were positively correlated among treatments (Fig.1).The analysis of variance revealed highly significant differences in genotype, environment, and genotype-environment interactions for the leaf wilting index (Table 1).The heritability of leaf wilting index was at least 96%under every treatment in either population.
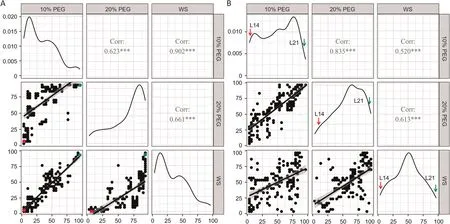
Fig.1.Correlations of leaf wilting index between 10%PEG,20%PEG and water-stress(WS)treatments applied to 188 soybean accessions(A)and 178 recombinant inbred lines(B).Plots on the main diagonals show the trait distributions under the three treatments.Scatterplots are displayed below the diagonal, and correlation coefficients and significance are shown above the diagonal.***, different at P < 0.001.Red and green dots or arrows indicate L14 and L21, respectively.
3.2.GWAS identified SNPs associated with leaf wilting index
Based on the cross-validation error rate and K-values obtained from the ADMIXTURE analysis of the 188 accessions (Fig.S1A),the panel was divided into seven main clusters (Fig.S1B).The mean LD decay distance was 178.7 kb when the threshold value of r2was set to 0.3 (Fig.S1C).A set of 26 SNPs were significantly associated with leaf wilting index: 13 under 10% PEG treatment,6 under 20% PEG treatment, and 7 under WS (Table 2).Significant markers were visualized with Manhattan plots,while important pvalue distributions were revealed by quantile–quantile(Q-Q)plots(Fig.2).SNPs rs022331 and rs022332 on chromosome 2 were identified under all three treatments and accounted for 13%–15% of phenotypic variation.SNP rs022333, detected under two treatments and explaining 13%–14% of phenotypic variation, was located in the same 248-kb LD block as the other two (Fig.S3).Three further SNPs: rs022331, rs022332 and rs022333, were detected under all three treatments by CMLM (Fig.S4).
3.3.Linkage mapping and QTL analysis
We further used linkage mapping in 178 RILs derived from a cross between L14 and L21.For the construction of linkage maps,6167 SNPs covering the 20 chromosomes were retained after removing SNPs with > 10% missing data or segregation distortions(Fig.S2).A total of 19 QTL that controlled leaf wilting index were detected in the individual environment, which included seven QTL under 10%PEG treatment,eight QTL under 20%PEG treatment,and four QTL under WS treatment,explaining 50%,48%,and 26%of the phenotypic variation,respectively(Table 3).The QTL qSW2 was detected under all three treatments, with the L14 allele associated with reduced leaf wilting index.This QTL was also detected in a joint analysis using the ICIM-ADD mapping method in the MET module, explaining 23.4% of the total phenotypic variation(Table S4).qSW2 was flanked by SNPs rs022314 and rs022331 on chromosome 2, and its interval was overlapped with the LD interval described above (Fig.S3).
3.4.Fine mapping of qSW2 and identifying candidate genes
qSW2 was detected in a 516-kb interval on chromosome 2(Fig.3A).To obtain a candidate genomic region for the positioningof qSW2,seven key informative recombinants were identified from 3526 BC2F3plants across the qSW2 target region using eleven KASP markers (Fig.3B).In comparison with the other recombinant homozygotes, RT5, RT6 and RT7 showed SW traits and higher RWC under WS conditions (Fig.3C).Finally, qSW2 was confined to a genomic interval of 60.24 kb bounded by markers KASP8 and KASP10 (Fig.3D).
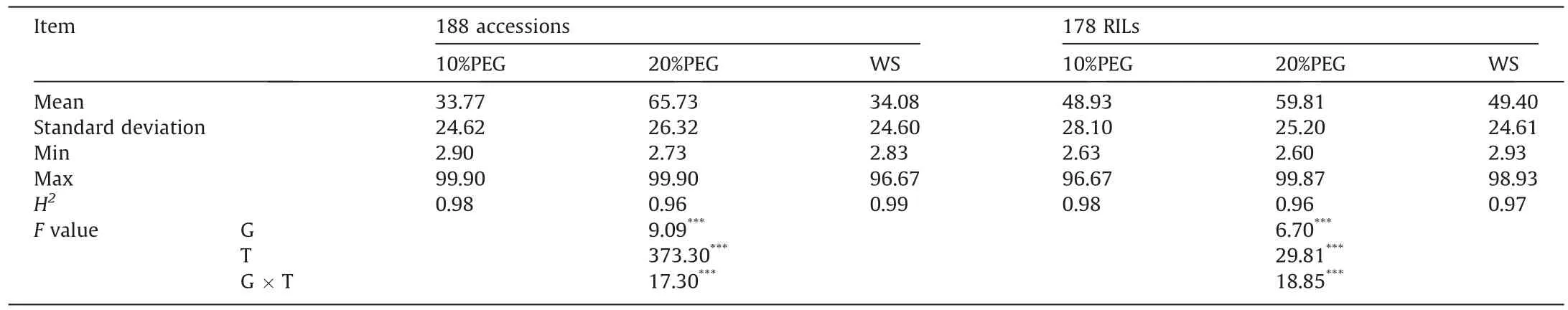
Table 1 Descriptive statistics and variance parameters estimated for leaf wilting index in 188 soybean accessions and 178 recombinant inbred lines(RILs)evaluated under 10%PEG, 20%PEG, and water-stress (WS) treatments.
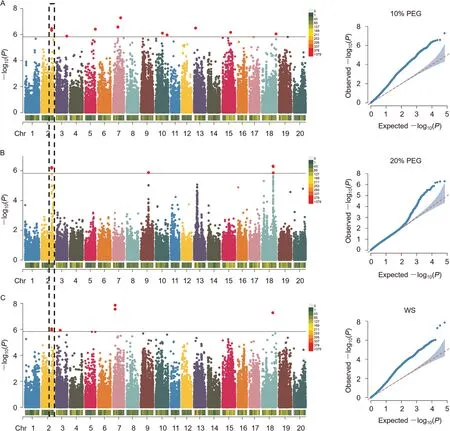
Fig.2.Manhattan and QQ plots for leaf wilting index under 10%PEG (A), 20%PEG (B) and water-stressed (WS) (C) treatments.The P-values correspond to the significance threshold of 1.47E-06.
According to the Glycine max reference genome database(https://www.soybase.org), this region contains three annotated genes: Glyma.02 g217900, Glyma.02 g218000, and Glyma.02 g218100.The first two encode respectively an uncharacterized protein At5g65660-like and an unknown protein, whereas the third encodes indole-3-acetic acid inducible 9 (AUX/IAA9).In 24-h post-treatment leaves, only this gene was differentially expressed between 20% PEG and well-watered conditions(Fig.3E).Glyma.02 g218100 expression was downregulated by 20%PEG treatment in L21, but showed nonsignificant changes in L14 and R26.The L14 and L21 DNA sequences of Glyma.02 g218100 harbored three SNPs and two InDels in the upstream region and one SNP in the downstream region (Fig.3F).
4.Discussion
A total of 38 QTL associated with SW have been registered in Soybase (https://www.soybase.org).Among these, only six QTL on chromosomes 2, 3, 6, 11, 17, 19 were stably expressed in multiple genetic backgrounds or environments[4,12,13].GWAS in theassociation panel detected 67 SNPs associated with SW, of which 18 were located within the linkage mapping interval in the previous studies [9,10].In the present study, GWAS revealed 26 SNPs associated with SW.Of these, three, all in the same LD block on chromosome 2, were detected under at least two treatments.To validate the efficacy of GWAS in gene discovery, 178 RILs derived from a cross between L14 and L21 were used for linkage mapping to identify a major-effect QTL qSW2 overlapping with the LD block on chromosome 2, evidenced to be shared on a SW-related QTL common region[14].There is a prevailing belief that MAS breeding could benefit from QTL detected in multiple environments as they are more stable than QTL with high effect values in a single environment [36].The QTL qSW2 was found to be stable across 10%PEG, 20%PEG and WS treatments, explaining 11.98%–24.71% of the total phenotypic variation, so it can be considered as a stable QTL for SW.
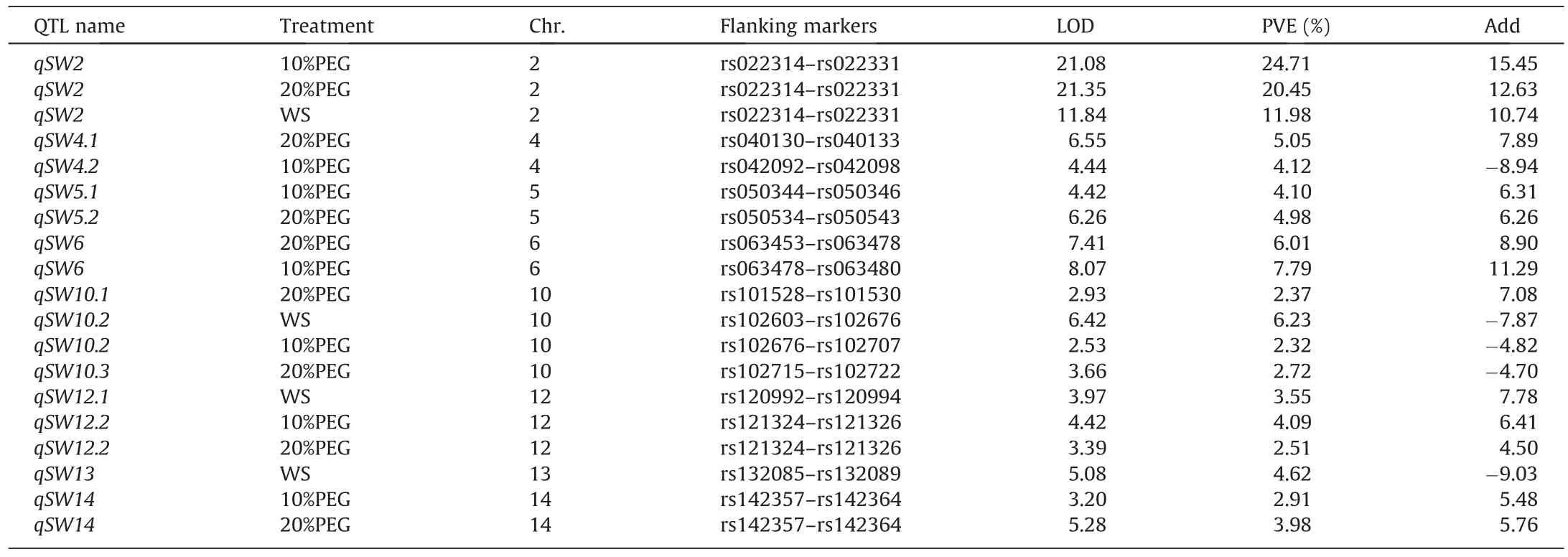
Table 3 QTL for leaf wilting index detected in recombinant inbred lines (RILs) evaluated under 10%PEG, 20%PEG and water-stressed (WS) treatments.
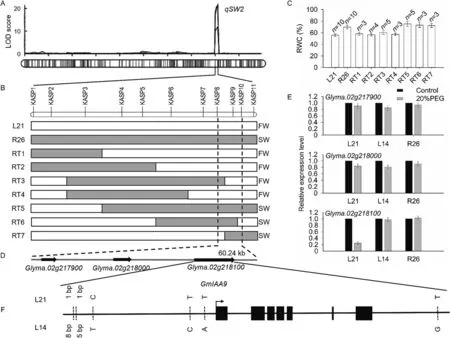
Fig.3.Fine mapping of qSW2 and identification of candidate genes.(A) The genetic region of qSW2 on chromosome 2.(B) Fine mapping of qSW2 by seven recombinants.Recombinant sites are marked with gray bars.White and gray boxes represent genotypes of L21 and R26,respectively.Target region of qSW2 is indicated by vertical dotted lines.SW,slow-wilting;FW,fast-wilting(C)Relative water content(RWC)of recombinants in the BC2F3 population under water stress.(D)Three candidate genes in the fine mapping region.(E) qRT-PCR expression levels for three candidate genes.(F) Upstream and downstream variation in Glyma.02 g218100 between L14 and L21.
To develop the BC2F3population for fine mapping of qSW2, the line R26,which had 54%genetic similarity to L21,was used as parent.With its uniplex SNP genotyping platform, KASP is capable of providing scalable flexibility to fine-map QTL with small to moderate numbers of markers at a relatively low cost[34].We identified seven key informative recombinants in BC2F3using eleven KASP markers in the target region.As these recombinants showed differing levels of leaf wilting as well as RWC,the qSW2 locus was delimited to a genomic span of 60.24 kb bounded by markers KASP8 and KASP10.
In the qSW2 interval, the gene Glyma.02 g218100 in soybean produces auxin transcription factor AUX/IAA9.This gene was down-regulated in L21, but not in L14 and R26, under 20% PEG treatment.This differential regulation is likely due to differences in the regulatory sequence of the gene, encompassing both the upstream and downstream regions.The plant auxin-responsive protein AUX/IAA functions as a transcriptional repressor and acts in the control of auxin signaling transduction by interacting with auxin-responsive factors (ARFs) [37].In the Arabidopsis genome,there are 29 AUX/IAA genes, among which IAA5, IAA6 and IAA19 have been identified as being regulated by the transcription factors DREB2A and DREB2B [38].They function in auxin signaling and respond to drought-induced stress [38].Their effect on leaf stomatal movement and plant drought tolerance is mediated by regulation of aliphatic sulfur-glycosome expression [39].The expression of the rice Aux/IAA gene OsIAA6 was upregulated in response to drought stress [40].Overexpression of the gene in transgenic rice increased drought tolerance, probably by regulating genes involved in auxin biosynthesis.OsIAA18 and OsIAA20, the AUX/IAA transcription factor genes of rice, also increase drought tolerance by regulating stress-induced ABA signaling [41,42].IAA9, a member of the AUX/IAA family of auxin-responsive proteins,functions in the regulation of fruit development and leaf morphology in tomato [43,44].Abiotic stresses imposed by NaCl, PEG and ABA inhibited IAA9 expression in leaves of drought-tolerant chickpea[45].Given the function of the Aux/IAA gene families,it is implied that Glyma.02 g218100 (GmIAA9) is a candidate gene for SW in soybean, which however needs further functional validation.
Known QTL for SW have been contributed mostly by PI 416937 and Jackson, although fast-wilting parents still provided donor alleles at a few loci [15].In this study, the favorable allele of qSW2 was contributed by L14, which not only was categorized as a drought-tolerant cultivar [46], but also produced the highest yield records, 4908 kg ha-1in Northeast China [47].In general,drought tolerance in soybean must involve traits that increase yield stability rather than simply ensuring survival [7,15,48].It has been found [4] that genotypes with SW traits yielded more than those with FW traits under drought stress.GmIAA9, a candidate gene for an SW QTL identified in a cultivar with SW and high yield, may find application in soybean breeding.
5.Conclusions
For leaf wilting index at seedling stage, a major-effect soybean QTL qSW2 was expressed under all water-stress treatments applied.The gene Glyma.02 g218100, encoding indole-3-acetic acid inducible 9, an AUX/IAA transcription factor, was assigned as a candidate gene for this SW QTL based on variations in its transcriptional expression and regulatory sequence.Our results provided a better understanding of the genetic architecture for SW and the cloning of the qSW2 will facilitate improving droughtresistance in soybean.
CRediT authorship contribution statement
Shengyou Li:Investigation, Data curation, Funding acquisition,Writing–original draft.Changling Wang:Methodology,Software,Validation.Chunjuan Yan:Investigation.Xugang Sun:Investigation, Methodology.Lijun Zhang:Investigation.Yongqiang Cao:Formal analysis, Funding acquisition.Wenbin Wang:Writing –review & editing.Shuhong Song:Funding acquisition, Project administration, Supervision.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The study was supported by the National Natural Science Foundation of China(32101795,32301782),National Key Research and Development Program of China (2016YFD0100201-01), Liaoning Provincial Major Special Project of Agricultural Science and Technology (2022JH1/10200002, 2021JH1/10400038), Key Research and Development Plan of Liaoning Science and Technology Department(2021JH2/1020027),and Shenyang Seed Industry Innovation Project (22-318-2-12).
Appendix A.Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2023.10.013.
杂志排行

The Crop Journal的其它文章
- Flowering-time regulation by the circadian clock: From Arabidopsis to crops
- Global characterization of OsPIP aquaporins reveals that the H2O2 transporter OsPIP2;6 increases resistance to rice blast
- Drought-triggered repression of miR166 promotes drought tolerance in soybean
- The OsBSK1-2-MAPK module regulates blast resistance in rice
- Natural variation of an autophagy-family gene among rice subspecies affects grain size and weight
- Rice gene OsUGT75A regulates seedling emergence under deep-sowing conditions