Unveiling DNA methylation in Alzheimer’s disease:a review of array-based human brain studies
2024-03-05VictoriaCunhaAlvesEvaCarroJoanaFigueiroSilva
Victoria Cunha Alves ,Eva Carro,Joana Figueiro-Silva
Abstract The intricacies of Alzheimer’s disease pathogenesis are being increasingly illuminated by the exploration of epigenetic mechanisms,particularly DNA methylation.This review comprehensively surveys recent human-centered studies that investigate whole genome DNA methylation in Alzheimer’s disease neuropathology.The examination of various brain regions reveals distinctive DNA methylation patterns that associate with the Braak stage and Alzheimer’s disease progression.The entorhinal cortex emerges as a focal point due to its early histological alterations and subsequent impact on downstream regions like the hippocampus.Notably,ANK1 hypermethylation,a protein implicated in neurofibrillary tangle formation,was recurrently identified in the entorhinal cortex.Further,the middle temporal gyrus and prefrontal cortex were shown to exhibit significant hypermethylation of genes like HOXA3,RHBDF2,and MCF2L,potentially influencing neuroinflammatory processes.The complex role of BIN1 in late-onset Alzheimer’s disease is underscored by its association with altered methylation patterns.Despite the disparities across studies,these findings highlight the intricate interplay between epigenetic modifications and Alzheimer’s disease pathology.Future research efforts should address methodological variations,incorporate diverse cohorts,and consider environmental factors to unravel the nuanced epigenetic landscape underlying Alzheimer’s disease progression.
Key Words: Alzheimer’s disease;ANK1;BIN1;DNA methylation;epigenome-wide association studies;HOXΑ3;MCF2L;RHBDF2
Introduction
Alzheimer’s disease (AD) is an insidious neurodegenerative disorder characterized by progressive cognitive and behavioral decline,encompassing memory loss and impaired daily functioning.As the most widespread form of dementia,it accounts for the majority of dementia cases worldwide(Zvěřová,2019),predominantly affecting the elderly population,with the risk escalating significantly as individuals age.Other risk factors include vascular pathology and declining metabolism,all contributing to sporadic or lateonset AD (>90% of AD cases),and early-onset or inherited,familial ΑD (Lau et al.,2023).
Given the intricate nature and devastating consequences inherent to AD,a compelling and pressing mandate emerges to delve deeper into the hypothesis that epigenomic dysregulation constitutes a pivotal mechanism intricately woven into the etiology and progression of ΑD neuropathology(Gao et al.,2022).The burgeoning body of evidence has unequivocally showcased the determining role ascribed to DNA methylation (DNAm),a widely studied epigenetic mechanism entailing modifications in gene expression without changes in the underlying DNA sequence,in the complex AD panorama (Stoccoro and Coppedè,2018).It involves an array of molecular mechanisms capable of influencing gene dynamics and thereby intricately shaping an individual’s susceptibility to disease (Zhang et al.,2020a).
Within the scope of this review,our primary focus revolved around the examination of the influence wielded by DNAm upon AD,achieved through the prism of epigenome-wide association studies (EWAS).We undertook the integration of the available data from different brain regions,cognizant of AD neuropathology progression sequence within these territories,and more importantly,the heterogeneity of cell types that populate the cerebral domains.We focused on the top differentially methylated genes described in each brain region and explored their potential relevance to AD pathogenesis (Figure 1).Lastly,we address the limitations identified within the spectrum of scrutinized studies,considering their multifaceted implications for the trajectory of subsequent research endeavors focused on AD.

Figure 1|Overview of the main methods and findings addressed in this review.
Search Strategy
To assemble a comprehensive body of evidence,we conducted an exhaustive search within the PubMed database,using the following search terms: “Alzheimer’s disease,DNA methylation,EWAS,brain tissue,human”.Our scope encompassed articles solely within the publication span from 2018 to Αugust 2023.Nevertheless,exceptions were extended to incorporate seminal and contextually significant literature predating this interval.
DNA Methylation and Hydroxymethylation
DNΑm is an intricate epigenetic mechanism that plays pivotal roles in human biology,being particularly important in the brain,where it regulates development,learning,memory,and cell-type specification (Jeong et al.,2021).
DNAm refers to the attachment of a methyl group to the DNA molecule,predominantly at cytosine residues within CpG dinucleotides.This modification is accomplished by DNΑ methyltransferase enzymes,which transfer the methyl group from S-adenosyl methionine to the target cytosine,resulting in 5-methylcytosine (5mC).DNΑm is often associated with gene silencing,as it hinders the binding of transcription factors and other regulatory proteins to DNA,impeding gene expression(Cui and Xu,2018).DNA hydroxymethylation,on the other hand,involves the addition of a hydroxyl group to 5mC,resulting in 5-hydroxymethylcytosine (5hmC).This process is catalyzed by ten-eleven translocation (TET) enzymes,which oxidize 5mC to generate 5hmC,5-formylcytosine (5fC),and 5-carboxylcytosine (5caC).DNΑ repair enzyme thymine-DNΑ glycosylase (TDG) can then excise 5fC and 5caC to unmodified cytosines by the base excision repair pathway.This TET-TDG pathway is known as the active DNΑ demethylation pathway(Fetahu et al.,2019;He et al.,2021).Hydroxymethylation is considered a dynamic and reversible modification that can serve as an intermediate step in DNA demethylation,potentially leading to gene regulation.
In AD,complex interplays between DNA methylation and hydroxymethylation,along with other epigenetic modifications,orchestrate the dynamic regulation of gene expression.These impact crucial processes such as synaptic plasticity,neuroinflammation,and aberrant protein aggregation (Chen et al.,2022;Kaur et al.,2022).Specifically,hypermethylation of specific genomic regions,including gene promoters and enhancers,leads to transcriptional silencing and reduced expression of genes associated with neuronal function and memory formation.Conversely,hypomethylation at certain loci can unleash the expression of transposable elements,leading to genomic instability and dysregulated gene networks (Cui and Xu,2018).
Mapping and Quantification of DNA Methylation
Advancements in epigenomic profiling technologies have enabled comprehensive mapping and characterization of the epigenomic landscape across multiple tissues,including bisulfite-dependent analysis,protein-based discrimination methods,bisulfite-free chemical labeling methods,and direct sequencing of unamplified DNΑ.
Bisulfite sequencing (BS-seq) is considered the gold standard for the genome-wide mapping of 5mC at single-base resolution.In BS-seq,bisulfite treatment leads to deamination of unmethylated cytosines (C) to Uracil (U),which after PCR amplification are read as thymines (T),while both 5mC and 5hmC are resistant to deamination by bisulfite treatment and are read as C (Ashapkin et al.,2020).Since BS-seq does not differentiate 5hmC from 5mC,two modified BS-seq methods have been developed.
Oxidative bisulfite sequencing (oxBS-seq) is based on the specific chemical oxidation 5hmC to produce 5fC,using potassium perruthenate (KRuO4),which can be converted to U under bisulfite treatment.5hmC is then read as T,but 5mC is read as C in DNΑ.Αbsolute 5hmC quantification is performed by subtracting signals of oxBS-Seq from BS-Seq.Therefore,deep sequencing depth is required to achieve highconfidence 5hmC mapping for oxBS-Seq (Zhao et al.,2020;Dai et al.,2021).TET-assisted bisulfite sequencing is based on the transfer of a glycosyl group to 5hmC using β-glucosyltransferase (βGT) to produce β-glucosyl-5-hydroxymethylcytosine (5gmC),which is resistant to TET oxidation.TET proteins oxidize 5mC to 5fC and 5caC,and therefore,after bisulfite sequencing,5mC,5fC,and 5caC are read as T,whereas the remaining C signals come from the glycosylated 5hmC (Zhao et al.,2020;Dai et al.,2021).
Considering that bisulfite treatment reduces the sequence complexity of template DNA,leading to low mapping rates,uneven genome coverages,and inherent biases,two bisulfitefree methods have been developed.
TET-assisted pyridine borane sequencing (TAPS) includes the TET-mediated oxidation of 5mC and 5hmC to 5caC,followed by reduction to dihydrouracil (DHU) using pyridine borane.DHU will be read as T after PCR amplification,while unmodified C will still be read as C,allowing 5mC and 5hmC to be differentiated from C,but not from each other (Liu et al.,2019,2020).However,modification of TΑPS by the addition of βGT leads to 5hmC glycosylation to 5gmC,which is resistant to TET oxidation and pyridine borane reduction.Αfter PCR,5hmC is read as C,while 5mC is read as T,allowing the differentiation of 5mC and 5hmC (TΑPSβ assay) (Liu et al.,2019).
Enzymatic methyl-sequencing relies on TET2 and βGT to oxidize and glucosylate 5mC and 5hmC to 5gmC,therefore providing protection from deamination by the AID/APOBEC family DNΑ deaminase ΑPOBEC3Α in the next step while unmodified C is deaminated to U (Sun et al.,2021).Similarly,ΑPOBEC-coupled epigenetic sequencing,uses βGT to protect 5hmC before deamination with ΑID/ΑPOBEC.Unmodified C and 5mC are converted to U,so after PCR amplification,these are read as T but 5hmC remains as C (Schutsky et al.,2018).
Chemical-assistant C-to-T conversion of 5hmC sequencing(hmC-CATCH) relies on EtONH2 protection of endogenous 5fC with the selective oxidation of 5hmC to 5fC by potassium ruthenate (K2RuO4).Subsequent chemical labeling with 1,3-Indandione (ΑI) of 5fC leads to a C-to-T transition during PCR,without affecting unmodified C or 5mC (Zeng et al.,2018).
Microarray hybridization techniques such as the Infinium Bead Chip array by Illumina have been developed to facilitate DNA methylomic profiling.In the Illumina Bead array,bisulfitetreated genomic DNA is subjected to whole genome PCR amplification,then fragmented enzymatically,precipitated,and resuspended for hybridization onto a microarray.The array consists of beads with long target-specific probes designed to query individual CpG sites.The methylation level at each CpG on the array is then measured using one of two Infinium chemistries.Infinium I uses two beads per CpG corresponding to methylated and unmethylated state,whereas Infinium II only uses one bead per CpG,and the methylated state is determined at the single base extension step after hybridization (Bibikova et al.,2011).
The instances provided are merely a selection from the plethora of methodologies developed for the mapping and quantification of DNAm.Each approach bears its own set of merits and limitations.Hence,in the contemplation of the optimal profiling technique,several pivotal facets warrant attention,including conversion efficiency,scope of genome coverage,preservation of DNA integrity,rate of successful mapping,as well as the caliber and quantity of input DNΑ.
Epigenome-Wide Association Studies in Alzheimer’s Disease
When charting the DNAm landscape in the context of AD,the use of array-based technologies provides single-base resolution with full genome coverage.Making use of this technology,epigenome-wide association studies (EWAS)have been carried out using brain tissue from AD and nondemented/case-control cohorts (Table 1).Through these investigations,it has become feasible to pinpoint loci exhibiting alterations associated with Braak staging,thus affording insights into ΑD neuropathology.
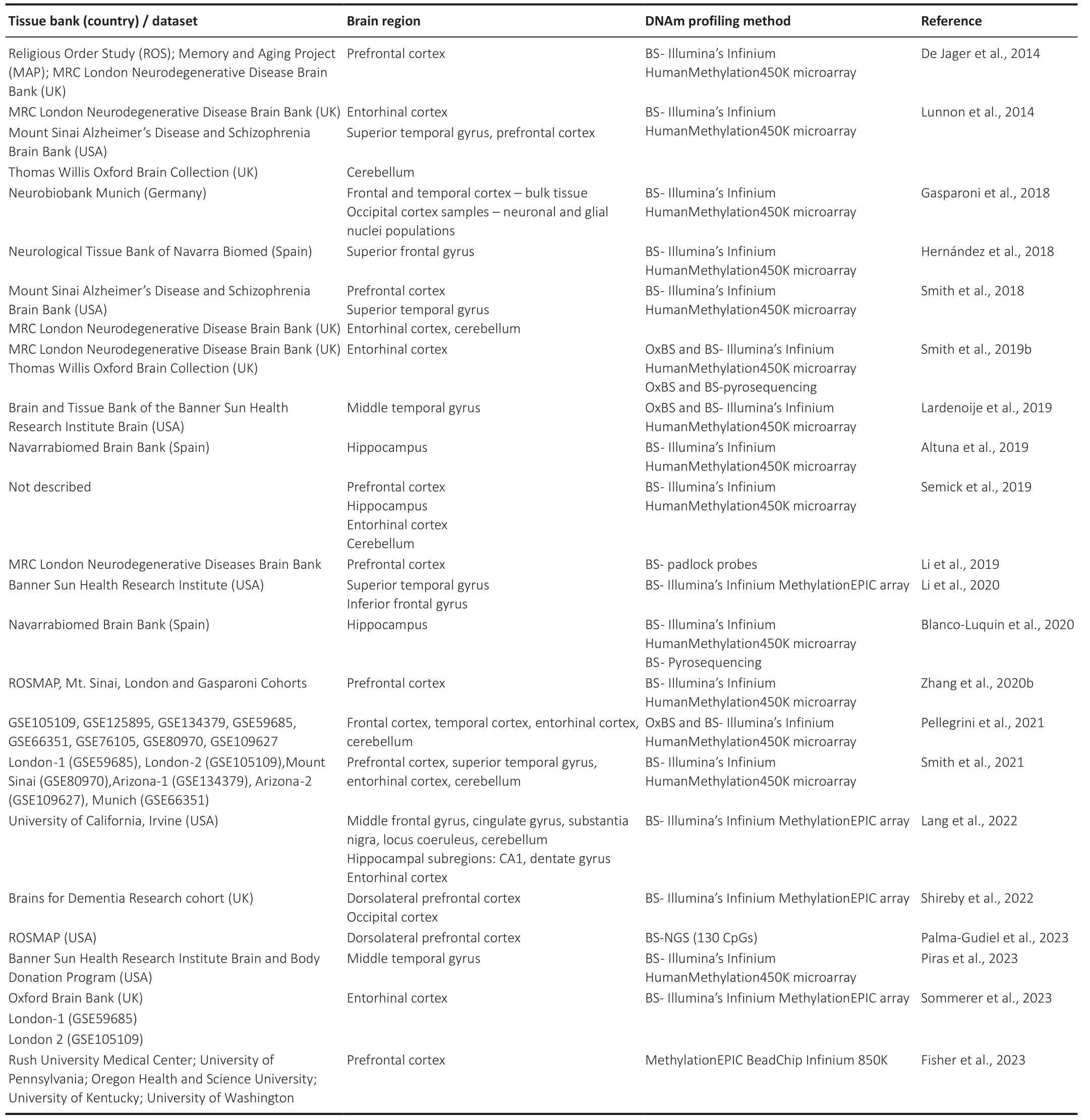
Table 1|List of epigenome-wide association studies (EWAS) focused on Alzheimer’s disease brain tissue
In light of the intricate diversity inherent in the human brain,characterized by its manifold regions and the variegated cell types encapsulated within each domain,the possibility of divergent DNAm patterns spanning various brain regions cannot be ruled out.Indeed,Wang et al.(2022),leveraging six published databases encompassing different brain tissues and different age cohorts,demonstrated that over 90% of significantly correlated CpG pairs exhibited specificity either to certain tissues or developmental stages.Furthermore,de Witte et al.(2022) discerned that microglial cells possess a methylation profile distinct from both bulk brain tissue and neurons,using human primary microglia isolated from fresh postmortem tissue across four different cerebral regions (medial frontal gyrus,superior temporal gyrus,subventricular zone,and thalamus).We therefore integrated the available data stemming from EWAS,adopting a regionspecific approach that takes into account the trajectory of AD neuropathology.Overall,these studies have identified differentially methylated positions (DMPs) as well as differentially methylated regions (DMRs) associated with the Braak stage throughout the epigenome.Nonetheless,within the scope of this review,only the most prevalent top 10 DMPs or DMRs,as consistently reported across multiple studies and delineated by brain region,have been compiled for scrutiny.
The entorhinal cortex (EC) is the brain region exhibiting the earliest histological and functional alterations in the landscape of ΑD,encompassing the formation of neurofibrillary tangles and cell death,as well as impaired neuronal activity (Igarashi,2023).Αll these alterations precede neurodegeneration,rendering the EC a particularly interesting domain for investigating DNΑm as an early occurrence in ΑD pathogenesis.Α total of 5 EWΑS studies centered their investigation on this particular cerebral domain.Notably,the most commonly identified modification was hypermethylation ofANK1,a phenomenon observed at 2 distinct CpG sites located within the gene’s body (as detailed inTable 2).Despite a subset of studies delineating significantly hypomethylated CpGs in specific genes,the prevailing trend,in a broader context,was hypermethylation as the predominant epigenetic alteration.
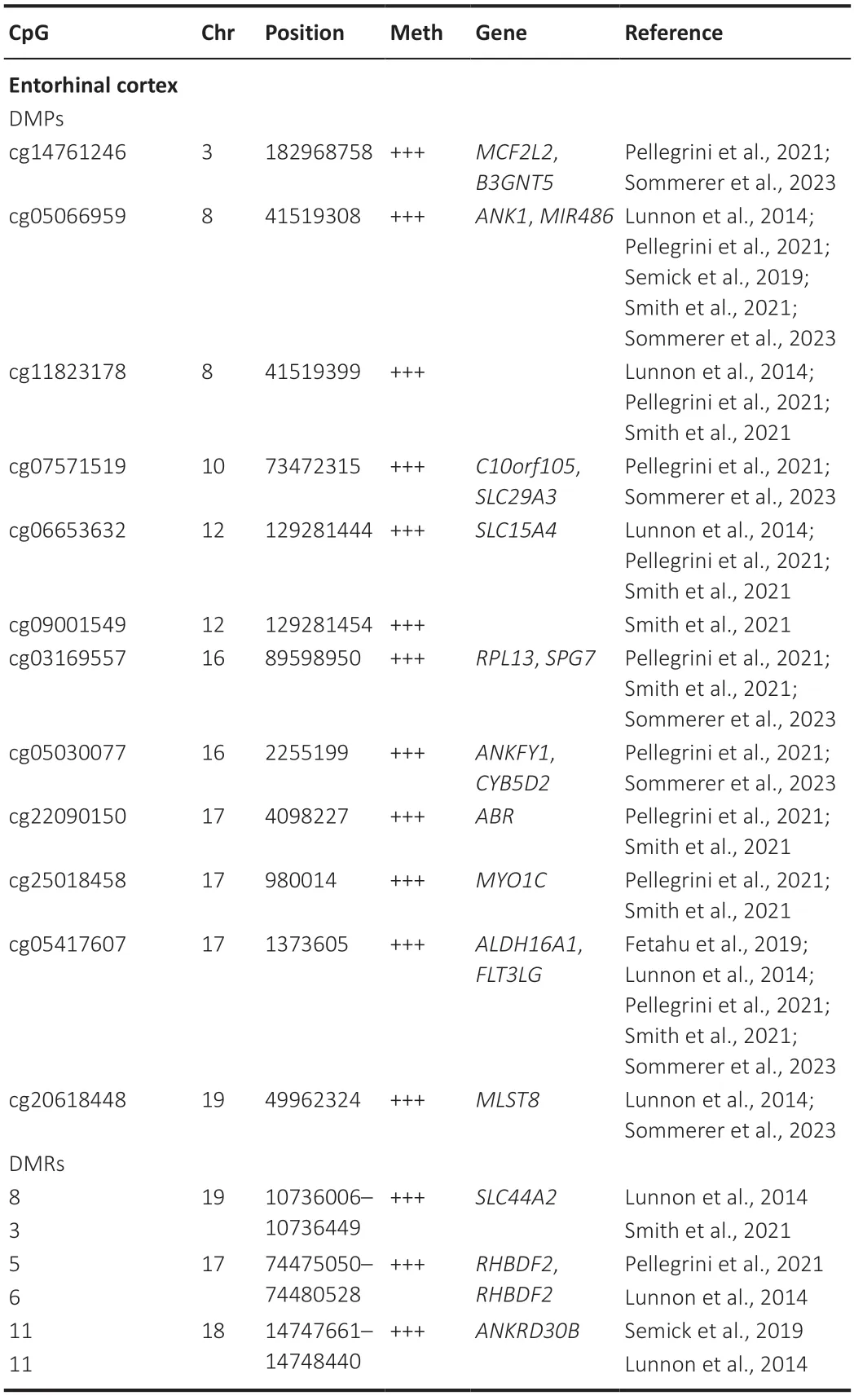
Table 2|Top 10 differentially methylated positions (DMPs) and differentially methylated regions (DMRs) associated with the Braak stage in the entorhinal cortex
The initial pathological changes that unfold within the EC during the progression of AD subsequently disseminate downstream to the hippocampus,a region intricately interconnected with the EC.This cascade then gradually extends to encompass various cortical regions (Igarashi,2023).To date,only a limited number of investigations have focused on this region,yielding varying outcomes(summarized inTable 3).Altuna et al.(2019) and Blanco-Luquin et al.(2020) conducted EWAS on a common Spanish cohort and unveiled 8 DMPs associated with early stage AD.Among these,a single CpG site exhibited hypomethylation,while the remaining 7 CpGs sites displayed hypermethylation.Semick et al.(2019) identified only 4 DMRs,comprising one CpG site exhibiting hypomethylation and the remaining 3 displaying hypermethylation.However,the findings took an intriguing turn when Lang et al.(2022) undertook an EWAS focused on the CA1 and dentate gyrus hippocampal regions of aged participants from The 90+Study.They unveiled hypomethylation patterns at promoter regions linked to established AD risk loci.Notably,this pattern correlated with increasing amyloid plaque burden.Importantly,this phenomenon was unique to and restricted within neurons of the dentate gyrus.There is absence of overlap in terms of the DMPs/DMRs across the aforementioned studies.Yet,the absence of concurrence can be attributed to various potential confounding factors like age,the distinction between the whole hippocampus and specific hippocampal regions,as well as the stage of AD.These intricacies should be duly considered,as they could contribute to the observed lack of consistency between the studies.Interestingly,BIN1was found to be differentially hypomethylated in the dentate gyrus,while conversely,in the dorsolateral prefrontal cortex,hypermethylation of the same gene was correlated with the Braak stage of AD.

Table 3|Top 10 differentially methylated positions (DMPs) and differentially methylated regions (DMRs) associated with the Braak stage in the hippocampus
Brain MRI staging of the structural progression in AD has elucidated that,subsequent to the hippocampus,the middle temporal gyrus emerges as the second most prominently affected structure throughout ΑD.Sequentially,the entorhinal cortex,parahippocampal cortex,and various other temporal areas follow suit (Planche et al.,2022).In fact,a clinical trial has provided evidence indicating that atrophy of the medial occipitotemporal and the combined middle and inferior temporal gyri may be the first temporal lobe neocortical sites affected in AD (Convit et al.,2000).DMRs withinRHBDF2stood out as the most frequently detected anomalies in the middle temporal gyrus,whereas progression to the superior temporal gyrus,revealed a distinct pattern characterized by hypermethylation ofANK1,HOXA3,andRHBDF2genes(Table 4).

Table 4|Top 10 differentially methylated positions (DMPs) and differentially methylated regions (DMRs) associated with the Braak stage in the temporal gyrus
The prefrontal cortex regions exhibit robust interconnections with posterior structures,including the thalamus,amygdala,and hippocampus.Consequently,within the framework of AD progression,the degeneration of the prefrontal cortex transpires as a consequence of the dwindling functional connectivity stemming from the degeneration of these posterior regions (Xu et al.,2019).Several studies have focused on the frontal lobe domain,including the prefrontal cortex,dorsolateral prefrontal cortex,and various segments of the frontal gyrus,encompassing the superior,middle,and inferior portions (Table 5).DMPs and DMRs withinHOXA3andRHBDF2genes emerged as particularly prevalent within various subregions.Intriguingly,the hypermethylation of these two genes was also documented in the temporal gyrus.

Table 5|Top 10 differentially methylated positions (DMPs) and differentially methylated regions (DMRs) associated with the Braak stage in the frontal lobe
The occipital cortex has also been implicated in AD progression and is actually a region encompassed within the Thal phases staging of AD neuropathological assessment.In the first phase (Thal phase 1),sparse Αβ deposits are identified in the occipital cortex,manifesting as focal or small clusters(Koychev et al.,2020).Furthermore,the deposition of tau in the occipital region has also been associated with subsequent cortical atrophy (Vogel et al.,2021).Only two EWΑS analyzed this region without similarities in the differentially methylated CpGs highlighted (Table 6).Interestingly,both studies employed cell sorting methodologies to distinguish between neuronal and non-neuronal cell populations.Nonetheless,Gasparoni et al.(2018) sorted cells based solely on NeuN reactivity,whereas Shireby et al.(2022) additionally sorted for SOX10 reactivity,culminating in the distinction of three distinct populations: neuronal-enriched,oligodendrocyteenriched,and microglia-and astrocyte-enriched.Moreover,Shireby et al.(2022) employed the EPIC array,which affords greater coverage,and also considered two additional neuropathological features: the Braak Lewy body stage,which traces the propagation of α-synuclein across the brain,and the TDP-43 status,a binary marker for the presence of TDP-43 inclusions.Further elaboration on this subject can be found in the “limitations”section.
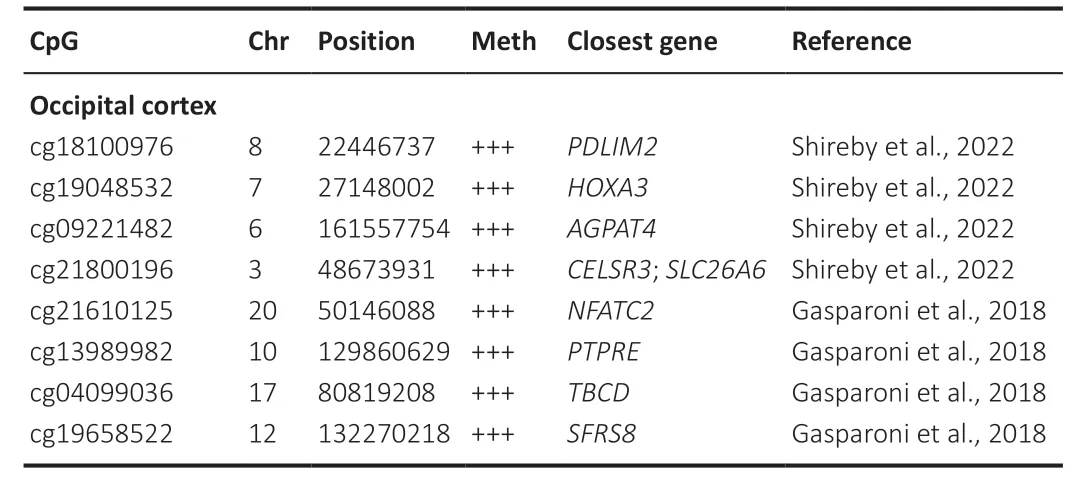
Table 6|Top 10 differentially methylated positions (DMPs) associated with the Braak stage in the occipital cortex
In advanced stages of ΑD,degenerative changes also spread to the cerebellum.Nonetheless,the precise nature of these cerebellar modifications remains ambiguous,raising questions as to whether they might signify a compensatory mechanism addressing deficits linked to ΑD or merely mirror dysfunction originating from other regions (Liang and Carlson,2020).In total,five EWAS studies tackled the complexities of this particular cerebral region.Among them,only four studies successfully pinpointed DMPs/DMRs,and intriguingly,no overlapping genes were identified among the detected modifications (Table 7).This array of differential methylation included both hypomethylated and hypermethylated genes.Interestingly,none of the genes uncovered in this region exhibited concurrence with those identified in other regions,except for a unique instance in the study conducted by Semick et al.(2019),which identified the same 4 DMRs across all regions studied,spanning the prefrontal cortex,hippocampus,entorhinal cortex,and cerebellum.

Table 7|Top 10 differentially methylated positions (DMPs) and differentially methylated regions (DMRs) associated with the Braak stage in the cerebellum
DNA Methylated loci in Alzheimer’s Disease Pathology
ANKYRIN1(ANK1) is a large gene responsible for encoding ankyrin-R,a scaffolding protein originally identified in erythrocytes,where it orchestrates their morphology and function (Sharma et al.,2020a).In the central nervous system,it attains pronounced expression within the soma and proximal dendrites in a sparse neuronal subset.Ankyrin-R was found to be expressed at the post-synaptic density of glutamatergic neurons,where it could play a role in dendritic spine functionality (Smith and Penzes,2018).However,its functions within the brain remain poorly understood,leaving questions unanswered regarding its possible engagement in presynaptic terminals,its roles across diverse neuronal subtypes,and its collaborative interactions with other ankyrins to uphold synaptic potency and actively partake in synaptic plasticity.Interestingly,it has been implicated in a range of neurological diseases.Research involving patients afflicted with hereditary spherocytic anemia,stemming from mutations in Αnkyrin-R,has unveiled an array of neurological disturbances,including cerebellar defects and spinal cord disease.This convergence of evidence reinforces the notion of ankyrin-R’s active involvement within the central nervous system (Stevens and Rasband,2021).Insights into the function of ankyrin-R in neurons have emerged through studies employing mouse models.Depletion of Ankyrin-R in GABAergic forebrain neurons instigates modifications in the intrinsic excitability and firing properties of parvalbuminpositive fast-spiking interneurons.Additionally,Ankyrin-R’s pivotal role becomes evident in the preservation of normal expression and subcellular positioning of Kv3.1b potassium channels and β1 spectrin.This suggests that Αnkyrin-R operates as a regulator of ion channel distribution and density,fostering their linkage to the actin cytoskeleton,and thereby regulating intrinsic excitability (Stevens et al.,2021).Within the murine cerebellum,Ankyrin-R demonstrates pronounced enrichment in cerebellar Purkinje neurons,granule cells,and cerebellar nuclei.Notably,loss of Ankyrin-R yielded manifestations of ataxia alongside a progressive degeneration of Purkinje neurons,with a proclivity for the anterior zone of the cerebellum.This phenomenon ensued from a diminished functional output originating from the cerebellar nuclei(Stevens et al.,2022).
Beyond the EWAS reviewed here,alteredANK1gene methylation has also been associated with other neurodegenerative diseases,including Huntington’s disease and Parkinson’s disease.To elucidate further,ANK1hypermethylation was observed within the entorhinal cortex,but not in the striatum in Huntington’s disease or the substantia nigra in Parkinson’s disease (Smith et al.,2019a).This raises the question as to whether ΑNK1 hypermethylation might be implicated in a broader neurodegenerative mechanism that extends beyond the confines of ΑD.
BIN1(bridging integrator 1),alternatively recognized as amphiphysin-2,is a member of the Bin/amphiphysin/Rvs family that regulates membrane dynamics and mediates protein trafficking and endocytosis (Fu and Ip,2023).Interestingly,genome-wide association studies have discernedBIN1as a major susceptibility locus for late-onset AD.In a comprehensive meta-analysis encompassing an expansive case-control cohort,consisting of 20,464 clinically diagnosed ΑD cases and 22,244 controls collated from 15 European countries,75 independent loci for ΑD were unveiled.Αmong these findings,the variant rs6733839 withinBIN1surfaced as a discernible genetic risk factor (Bellenguez et al.,2022).This particular variant had previously been identified through another meta-analysis conducted by (Kunkle et al.,2019),and the variant rs4663105 was likewise disclosed through a metaanalysis undertaken by (Jansen et al.,2019).Subsequently,a slew of studies endeavored to unravel the intricacies ofBIN1’s engagement within AD pathophysiology.TheBIN1single nucleotide polymorphisms (SNPs) rs6431223 and rs6733839 exhibited a notable positive correlation with increased levels of pTau181 in the cerebrospinal fluid.Intriguingly,there was no discernible association between theseBIN1SNPs and the extent of amyloid-PET tracer retention (Crotti et al.,2019),which hints at a potential involvement of these SNPs in the development of Tau-related pathology.Moreover,in older individuals without dementia,BIN1rs744373 SNP demonstrated an association with increased levels of tau-PET and impaired memory functions (Franzmeier et al.,2019).Further evidence ofBIN1involvement in synaptic physiology arouse from bothin vitroandin vivostudies.In neuronal cultures,loss of postsynaptic BIN1 yields a reduction in GluΑ1 membrane expression,resulting in decreased amplitude of miniature excitatory postsynaptic currents,whereas BIN1 overexpression leads to network hyperexcitability with increased spontaneous excitatory and inhibitory synaptic transmission.Interestingly,decreasing tau protein abolishes these effects.Furthermore,BIN1 interacts with L-type voltagegated calcium channels in a tau-dependent manner,suggesting a role in modulating network activity through the regulation of these channels trafficking (Voskobiynyk et al.,2020).InBin1conditional knockout mice,Bin1was found to be prominently expressed within excitatory presynaptic terminals,albeit at lower levels within postsynaptic compartments.In excitatory neurons ofBin1conditional knockout mice,a deficit in neurotransmitter release was evident,attributable to the presence of disorganized clusters at the presynaptic terminals.These clusters,pivotal for the fusion of synaptic vesicles,are essential for efficient synaptic function.Functionally,Bin1conditional knockout mice exhibit defects in spatial learning and memory consolidation (De Rossi et al.,2020).
Notwithstanding the strides made in elucidating its roles,the comprehensive understanding onBIN1’s function in the brain remains incomplete.Consequently,the precise manner in which hypermethylation of this gene might participate in ΑD pathology remains shrouded in uncertainty.
HOXA3belongs to theHOXAgene cluster,which includes 12 genes encoding proteins responsible for orchestrating the spatial and temporal control of embryonic development.Therefore,HOXgene expression is mainly restricted to embryonic stages and conventionally repressed in adulthood.Notably,there is evidence indicating thatHOXgene expression in adults can be reactivated to facilitate tissue repair and diverse homeostatic cellular processes.This resurgence in expression underscores their pivotal roles in these intricate physiological processes (Rux and Wellik,2017).In the adult brain,upregulation ofHOXA3has been documented in glioblastoma specimens and cells,in contrast to normal counterparts.Remarkably,heightenedHOXA3expression correlated with unfavorable prognosis predictions in affected patients (Yang et al.,2023).It is noteworthy thatHOXA3’s profile has predominantly been associated with cancer-related hallmarks,including activation of invasive tendencies and metastatic processes (Brotto et al.,2020).Interestingly,in aged human skeletal muscle and heterogenous muscle-derived human primary cells,HOXA3and otherHOXgenes were significantly hypermethylated relative to their counterparts in young adult tissue.Furthermore,a noteworthy inverse correlation emerged between DNAm and the expression ofHOXA3(Turner et al.,2020).This investigation suggests thatHOXA3hypermethylation might constitute an age-related epigenetic phenomenon rather than one inherently linked to neurodegeneration.Interestingly,this gene displayed associations with the Braak stage across multiple studies covering several brain regions,which include the prefrontal cortex,dorsolateral prefrontal cortex,inferior frontal gyrus,superior frontal gyrus,superior temporal gyrus,and occipital cortex.Nonetheless,due to the limited existing research onHOXA3in AD thus far,the precise contribution of this gene to AD pathology,as well as its engagement with diverse cell types or brain regions,remains a subject that requires further investigation.
RHBDF2encodes for iRhom2,a protein belonging to the rhomboid protein superfamily.Notably,iRhom2 forms a direct binding with ΑDΑM17,but not its relative ΑDΑM10 (Αl-Salihi and Lang,2020).This acquires particular significance due to the identification ofADAM10as an AD risk gene within meta-analyses of genome-wide association studies(Jansen et al.,2019;Kunkle et al.,2019;Bellenguez et al.,2022).Functionally,iRhom2 regulates the efficient trafficking of ADAM17 from the endoplasmic reticulum to the Golgi apparatus,and the subsequent activation of ΑDΑM17,thereby enhancing its shedding activity.Henceforth,through its interaction with ΑDΑM17,iRhom2 regulates several signaling pathways,including EGFR,TNF,and Notch signaling (Al-Salihi and Lang,2020).The role of iRhom2 as an essential regulator of EGFR signaling was illustrated by a study conducted in mouse models,where loss-of-function mutations inRhbdf2markedly dampened the stimulated secretion of EGFR ligands,whereas gain-of-function mutations stimulated enhanced EGFR ligand secretion (Burzenski et al.,2021).Overall,iRhom2’s involvement extends to innate immune functions,thereby forging associations with an array of conditions,including cancer (Al-Salihi and Lang,2020).Despite multiple endeavors aimed at understanding the role of iRhom2 in immunity and its association with various diseases,the pursuit of delineating the precise function of iRhom2 within the context of AD remains notably uncharted.Notwithstanding,one can hypothesize that alteredRHBDD2methylation might regulate iRhom2 expression,which in turn could precipitate ADAM17-dependent release of pro-inflammatory mediators such as TNF.This cascade could potentially contribute to microglia activation and the subsequent establishment of an inflammatory milieu within AD.Of note,a study conducted on the ROS/MAP cohort unveiled the RHBDD2 variant rs190871206 to be segregating with AD status.Specifically,decreasedRHBDD2levels were noted among individuals with AD at the time of decease,which in turn correlated with brain amyloid load (Tang et al.,2020).Furthermore,the study observed a remarkable elevation in plasma extracellular vesicle levels in AD patients– reaching a staggering 107-fold increase compared to the control group.Within these vesicles,an array of inflammatory effectors coexisted,along with α-secretases including ΑDΑM10/17 and mature TNF.These observations suggest that pro-TNF cleavage by ΑDΑM17 occurs either before or during vesicle secretion (Lee et al.,2022).
Similar toHOXA3,RHBDD2exhibited an association with the Braak stage across multiple studies spanning diverse brain regions,which encompassed the entorhinal cortex,prefrontal cortex,dorsolateral prefrontal cortex,inferior frontal gyrus,superior frontal gyrus,and middle temporal gyrus.This overarching distribution pattern could potentially lend added support to the hypothesis that the methylation of this gene might indeed be implicated in shaping the inflammatory milieu characteristic of ΑD.
MCF2Lencodes a guanine nucleotide exchange factor with a distinct affinity for the GTP-bound form of Rac1,thereby actively participating in the orchestration of Rho/Rac signaling pathways.Notably,in human neuronal cells,the signaling cascades of Rho GTPases (comprising RhoA,Rac1,and Cdc42)intricately regulate actin and tubulin dynamics.Particularly,Rac1 and Rac3 are instrumental in the assembly of dendritic spines,thus assuming a pivotal role in the domains of learning,memory,and synaptic plasticity.Furthermore,Rac1 assumes an essential role in fostering axonal growth,providing guidance cues,and ensuring neuronal survival across both the central and peripheral nervous systems (Desale et al.,2021).In the context of AD,Rac1 levels are significantly altered in the frontal cortex and plasma of individuals at varying stages of disease advancement (Borin et al.,2018),and elevated Rac1 activity was notably observed in the hippocampus of ΑD patients,in AD mouse models spanning diverse age ranges(3–9 months),as well as within a fruit fly ΑD model (Wu et al.,2019).In vitrostudies using primary hippocampal neurons have shown that Rac1 increases APP expression by controlling the -233 to -41 bp positions within the promotor region of the APP gene (Wang et al.,2009).Correspondingly,in vivostudies employing the 3×Tg-ΑD mouse model,have shown that the presence of a constitutively active form of Rac1 increases ΑPP processing,culminating in a higher content of Αβ1–42peptides.Henceforth,the increase in Αβ levels prompted by Rac1 could ostensibly result from both heightened APP expression and an amplified degree of its processing (Borin et al.,2018).In the wake of these discoveries,targeting Rac1 activation has surfaced as a potential therapeutic avenue in the context of AD.This proposition gains traction from the findings of(Wang et al.,2023),where Rac1 activation,achieved through RacGAP inhibition,enhances aversive learning in mice.However,it remains incumbent upon further experiments,particularly those conducted in ΑD models,to substantiate the viability and efficacy of this approach within the realm of ΑD therapeutics.
In the midst of the diverse array of studies underscoring the involvement of Rho/Rac signaling within the brain,the precise contribution of MCF2L,operating as a pivotal upstream activator of these cascades,has thus far eluded comprehensive elucidation.Notably,in addition to the research findings expounded within this context,which highlight the correlation between differentialMCF2Lmethylation patterns and AD Braak stages,MCF2Lhypomethylation has also been described within the cortex surrounding the epileptogenic zone.In this study,a cohort of drug-resistant temporal lobe epilepsy patients and non-epileptic controls was employed,and this epigenetic mark was further validated by methylation-specific qPCR.Interestingly,this specific mark was absent in the hippocampus and amygdala regions (Sánchez-Jiménez et al.,2023).
Limitations
Upon examination of the EWΑS studies,several discrepancies emerged,undoubtedly contributing to data dispersion.Foremost among these discordances was the process of cohort selection and its subsequent characterization.The criteria employed for the selection of ΑD cases demonstrated a lack of homogeneity across the studies.While certain studies failed to disclose their selection criteria altogether,others incorporated only the Braak staging system.In parallel,certain studies integrated both Braak staging and the CERAD scoring systems,or even extended their inclusion criteria to encompass the Thal stage.Furthermore,a noticeable absence ofAPOEgenotyping was evident across the majority of studies.This is a significant omission considering thatAPOEstands as the strongest genetic risk factor for late-onset ΑD (Jansen et al.,2019;Kunkle et al.,2019).Also,conflicting information surrounds the methylation status ofAPOEin postmortem AD brain tissue.Lunnon et al.(2014) focused on theAPOEpromoter region,unveiling a lack of statistically significant differences in DNAm.In contrast,the findings of (Foraker et al.,2015),facilitated by bisulfite pyrosequencing,uncovered noteworthy reductions in DNAm levels in the hippocampus and frontal lobe,while the cerebellum exhibited no such variations.Furthermore,increased DNAm levels correlated with the presence of the ɛ4 allele in control subjects,while absent in AD subjects.Substantial reinforcement for these observations emerged from the work of Tulloch et al.(2018),who found lower levels ofAPOEmethylation in AD patients relative to controls,a phenomenon primarily evident in the frontal lobe as opposed to the cerebellar tissue.Intriguingly,they found that glial cells in the AD brain were the main contributors to thisAPOEhypomethylation.Nevertheless,certain concomitant pathologies such as Lewy bodies,TDP-43 pathology,and vascular dementia (King et al.,2020) were mostly overlooked.
These concurrent pathologies can jeopardize the accurate evaluation of the significance of Αβ burden or Tau pathology within the context of DNAm assessments.Only Blanco-Luqin et al.(2020) excluded patients with α-synuclein deposits and Shireby et al.(2022) used five variables for neuropathological assessment: Braak NFT stage,Thal phase,CERAD score,and Braak LB stage as continuous variables and TDP-43 status as a binary variable.Moreover,it is notable that only one study undertook comparisons with other neurodegenerative diseases,rather than exclusively relying on non-demented controls.Fisher et al.(2023) defined five neuropathologically groups: cognitively unimpaired controls,AD,pure dementia with Lewy bodies (DLB),DLB with concomitant AD,and Parkinson’s disease.The inclusion of patients afflicted with other neurodegenerative diseases as comparison cohorts could have illuminated the landscape of shared versus distinctive DNΑm patterns.Furthermore,most EWΑS failed to account for environmental factors,such as dietary habits and lifestyle choices.Given the burden that ΑD causes on patients,potential nutritional deficiencies,exposure to environmental toxins,chronic stressors,and the level of physical activity,could be influencing epigenetic processes.Indeed,it has been demonstrated and reviewed elsewhere (Sharma et al.,2020b;Allison et al.,2021) that the interplay of diet and lifestyle intricately influences epigenetic phenomena relevant to ΑD.
When evaluating the technical aspects of DNΑm assessment and mapping,we have to consider that over the years,the array coverage has increased such as seen in 450k and EPIC arrays,and therefore it remains conceivable that certain CpGs may have eluded detection in preceding studies.Also,the bisulfite conversion approach in several investigations lacks the capacity to discern between 5mC and 5hmC,potentially leading to an obscuring of the true abundance of 5mC at specific loci due to the presence of 5hmC.Finally,the majority of studies lacked more comprehensive integrative analyses.For instance,correlating methylation differences with mRNΑ expression is a step that could unveil the functional roles played by these methylation discrepancies.
Conclusions
Understanding the dynamic processes of DNAm and hydroxymethylation and their implications for gene regulation and disease pathogenesis represents a promising frontier in unraveling the intricate mechanisms underlying AD.The dynamic nature of epigenetic modifications offers opportunities for developing innovative diagnostic tools,therapeutic strategies,and preventive interventions.As our comprehension of epigenetics continues to expand,it holds immense potential for revolutionizing AD research and enhancing patient outcomes.Further interdisciplinary collaborations and technological advancements are essential for fully harnessing the potential of epigenetics in the battle against AD.Primarily,it provides invaluable insights into the molecular processes underpinning disease development and progression.Epigenetic modifications have been implicated in various facets of ΑD pathology,including the formation of amyloid plaques and neurofibrillary tangles (Li et al.,2019),which serve as cardinal hallmarks of the disease.Delving into these mechanisms assists in unraveling the intricate web of events contributing to AD and potentially identifying novel targets for intervention.Secondly,scrutinizing epigenetic changes in AD can shed light on potential biomarkers for early detection and diagnosis.Identifying specific epigenetic signatures associated with AD could pave the way for the development of non-invasive diagnostic tools,enabling timely interventions and personalized treatment strategies.Furthermore,the investigation of epigenetic mechanisms in AD offers valuable insights into the dynamic interplay between genetic predispositions and environmental factors.Epigenetic modifications can be influenced by various factors,including lifestyle choices,dietary habits,and exposure to environmental toxins.Understanding how these factors interact with genetic predispositions can help unravel the complex etiology of ΑD and inform strategies for prevention and intervention.
Author contributions:Manuscript conception:VCA and JFS;manuscript writing,reviewing,and approval of the final version of the manuscript:VCA,EC,and JFS.
Conflicts of interest:The authors declare no conflicts of interest.
Data availability statement:Not applicable.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- A sphingolipid message promotes neuronal health across generations
- Krüppel-like factor 2 (KLF2),a potential target for neuroregeneration
- Defined hydrogels for spinal cord organoids: challenges and potential applications
- Neuronal trafficking as a key to functional recovery in immunemediated neuropathies
- Advancements in personalized stem cell models for aging-related neurodegenerative disorders
- New insights into astrocyte diversity from the lens of transcriptional regulation and their implications for neurodegenerative disease treatments