Materials Studio软件在计算化学和计算材料学课程教学中的应用
2024-03-04许真铭刘庆生陈江安
许真铭,刘庆生,陈江安
1 南京航空航天大学材料科学与技术学院,南京 210016
2 江西理工大学材料冶金化学学部,江西 赣州 341000
3 江西理工大学资源与环境工程学院,江西 赣州 341000
1 引言
立德树人是新时代高校教育的根本任务。如何培养好学生,同时提高学生理论知识水平和实践创新能力;如何在课程理论教学中融合现代信息化手段,在理论课程教学中强化基本方法原理和软件操作实践的联系,进一步提升课程教学质量效果,是高校教师课堂教学面临的新挑战和必须解决的问题。笔者长期从事新能源材料计算模拟研究工作,承担本单位计算化学、计算材料学等本科生、研究生专业课程。结合自己的科研工作实践,一直致力于将科研计算模拟案例分析和软件操作使用有机融合到量子化学计算、第一性原理计算、分子动力学模拟、蒙特卡洛模拟等理论方法教学过程,让理论知识概念点落脚于具体的计算案例和计算操作,使教学内容从抽象到直观,培养具备现代化材料理论和计算设计思维能力的复合型人才,实现科研反哺高质量教学。
2 理论基础与软件操作
计算化学、计算材料学等课程主要涉及量子化学计算、第一性原理计算、分子动力学模拟、蒙特卡洛模拟等理论方法的讲解,具有知识概念抽象、繁杂等特点[1,2]。量子化学计算是基于量子力学基本原理和Hartree-Fock等近似方法求解薛定谔方程,解析电子波函数,进而获得体系的基态性质[2];第一性原理计算是基于量子力学基本原理和密度泛函理论,不依赖任何经验参数,通过若干近似处理后求解体系薛定谔方程,进而得到电子结构以及材料所有的基态性质[3,4];分子动力学模拟是基于经典力学物理框架,通过求解体系牛顿运动方程,模拟体系的动力学演化过程,结合统计热力学原理,获得体系热力学和动力学输运性质[5];蒙特卡洛模拟是以概率统计理论方法为基础,通过随机抽样方法求解体系热力学和动力学性质[5]。
本文中,我们利用Materials Studio 2020软件配合量子化学计算、第一性原理计算、分子动力学模拟、蒙特卡洛模拟等四个理论方法章节中的部分教学内容进行锂电池材料模拟计算实践操作,并以图片形式对部分计算结果进行展示。Materials Studio软件是美国BIOVIA公司专为材料科学、化学领域开发的一款多尺度材料模拟计算软件平台,主要包括Visualizer、CASTEP、DMol3、Forcite、AmorphousCell、Sorption、QSAR、Morphology、Reflex等共计22个涵盖量子力学、经典分子力学、介观动力学、蒙特卡洛、定量构效关系、结构建模以及晶体结构精修解析等模块。如图1所示,Materials Studio软件可基于Windows操作系统运行,具有界面友好、易操作等特点,非常适合初学者入门计算模拟操作以及课堂教学使用。利用Materials Studio软件中Visualizer模块能够对分子、晶体、表面、高分子及介观结构的模型进行高效建模、复制和查看。同时,Visualizer还支持对所有模拟计算结果进行各种分析,可以输出文本数据和图表,可以对特定的计算模拟结果进行动画演示。
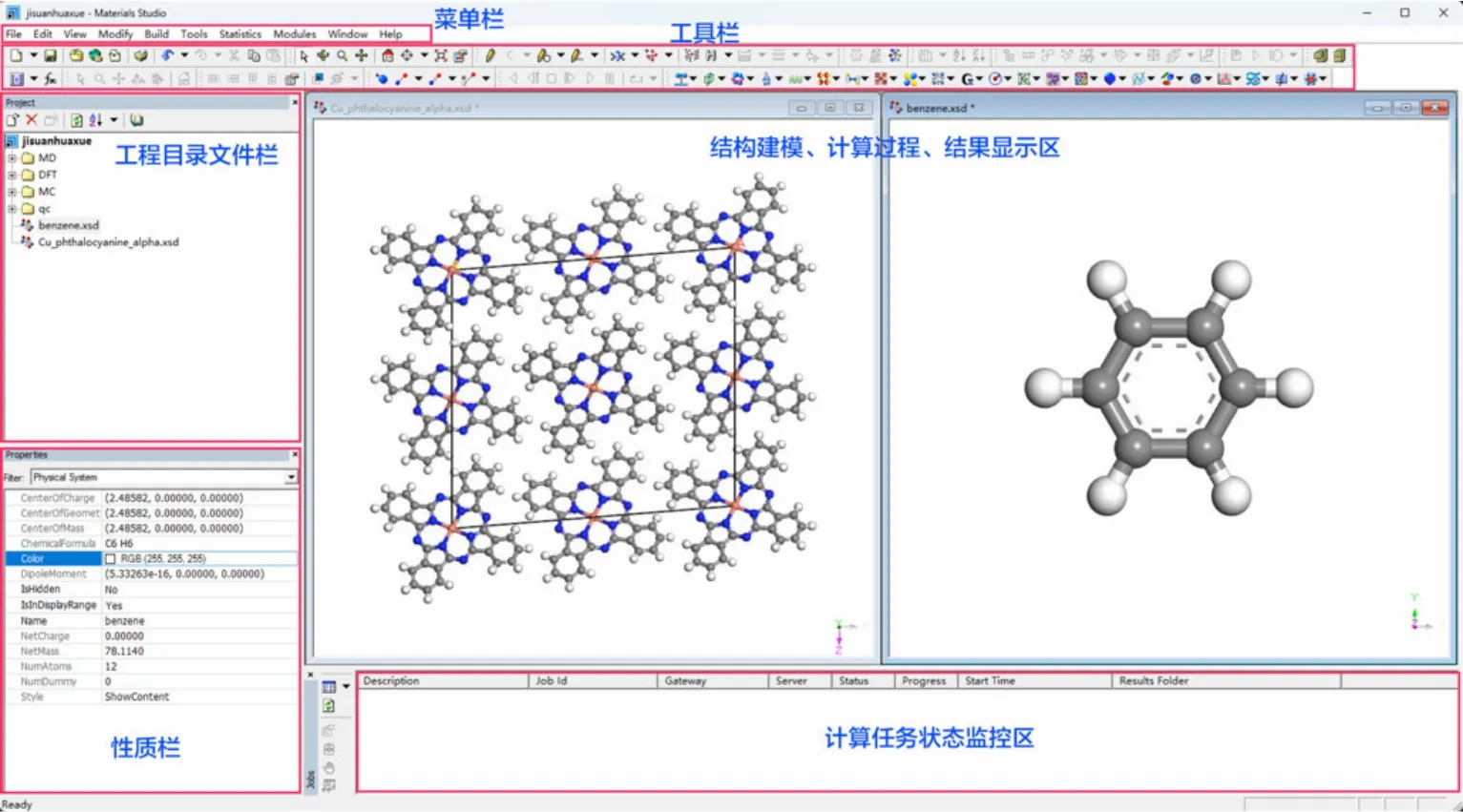
图1 Materials Studio多尺度材料模拟计算软件使用界面
3 计算模拟操作实践教学案例
3.1 锂离子电池有机电解液溶剂分子量子化学计算
锂离子电池有机电解液溶剂分子的基础(电)化学性质决定着电解液的综合电化学性能。目前,量子化学计算等计算模拟手段已经广泛地应用到电解液溶剂分子的研发过程[6]。在本单位计算化学课程的量子化学计算理论方法教学过程中,引入EC (碳酸乙烯酯)、EMC (碳酸甲乙酯)、DMC (碳酸二甲酯)等商业化锂离子电池电解液常用溶剂分子的电子结构量子化学计算案例及其DMol3[7]量子力学计算软件实践操作,帮助学生进一步巩固量子化学计算方法、计算思路以及核心物理概念。主要步骤如下:
(1) EC、EMC、DMC分子结构建模,并用Visualizer模块的clean功能进行初步优化(注意:clean功能是根据经验键长键角对结构进行合理化,非结构优化);
(2) 启动DMol3量子力学计算模块,选择Geometry Optimization任务,选择Fine计算精度,选择Hybrid-B3LYP泛函,勾选自旋非限制,使用All Electron处理芯电子,采用DNP基组,其他设置参数采用软件默认值,对第一步所获得的三种溶剂分子结构分别进行几何结构优化计算(优化原子坐标),待能量、力、位移均达到收敛值后,获得最优分子结构;
(3) 针对上一步所获得的最优分子结构,启动DMol3量子力学计算模块,选择Energy单点能计算任务,选择Ultra-fine计算精度,勾选电子态密度、电荷密度、静电势、电子轨道以及电子布居数等性质,其他量子化学计算设置参数与第二步相同,再次执行计算;
(4) 启动DMol3模块中的Analysis功能,分别执行Density of state、Electron density、Orbitals、Population analysis、Potentials分析,获得电子态密度、电荷密度、HOMO和LUMO、电子布居数以及静电势等电子性质数据,如图2所示。
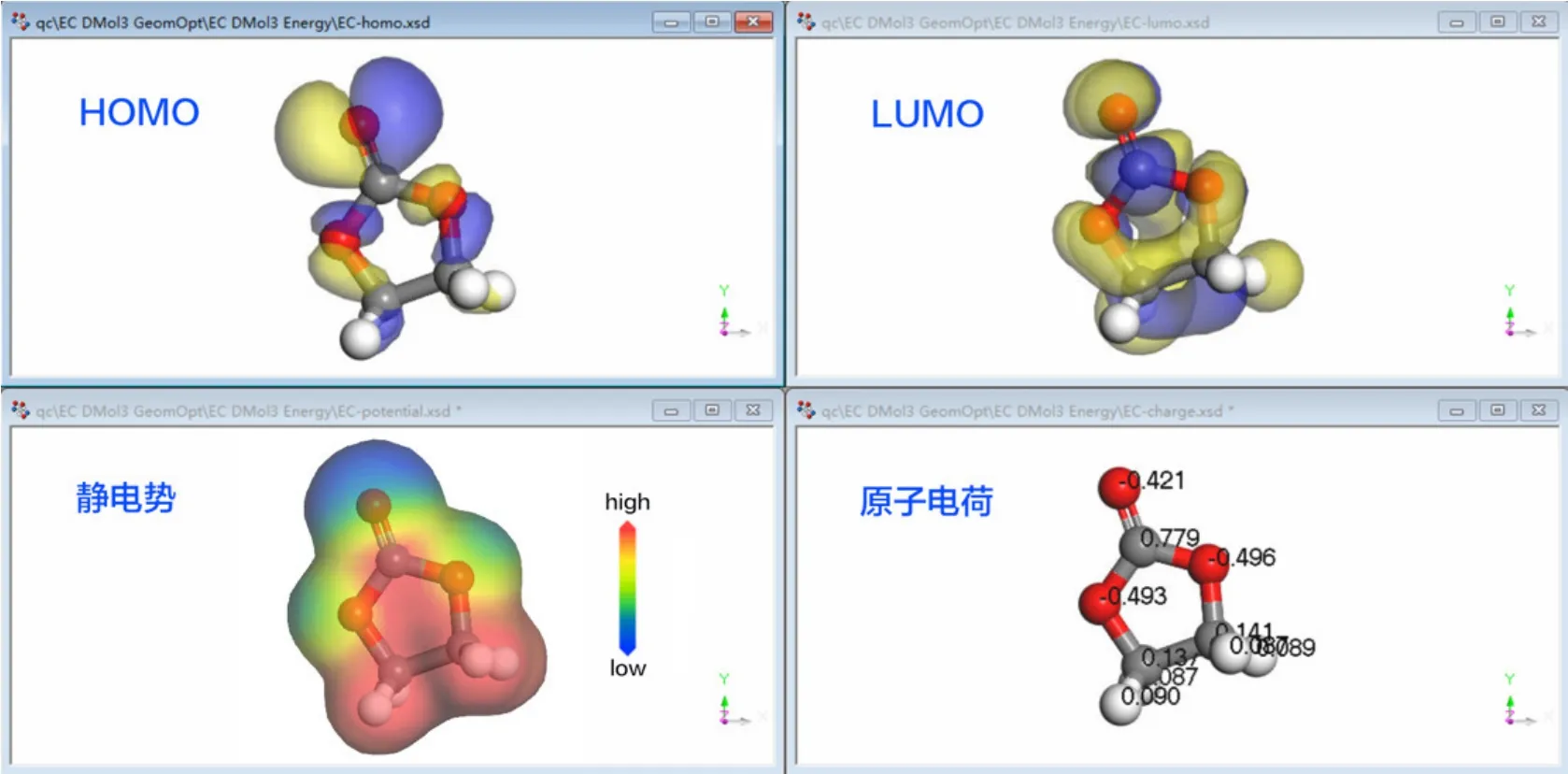
图2 EC溶剂分子量子化学计算结果
通过上述锂离子电池有机电解液EC、EMC、DMC溶剂分子计算案例教学,让学生掌握量子化学计算主要步骤,进一步掌握轨道基组、Hartree-Fock近似、自洽场等量子化学计算方法中的核心概念,掌握分子结构建模方法,掌握电子态密度、电荷密度、静电势、HOMO和LUMO电子轨道以及电子布居数等电子结构性质的计算方法。
3.2 锂离子电池正极材料LiFePO4第一性原理计算
LiFePO4是目前商业化锂离子电池的正极材料,具有高安全性、低成本等优势。但是,LiFePO4的本征电子导电性很差,因此在实际运用中需要进行包覆导电碳[8]。同时,部分实验和第一性原理计算研究表明合适的元素掺杂也可提高LiFePO4电子导电性[9,10]。在计算材料学课程的第一性原理计算理论方法教学过程中,引入LiFePO4电子结构(能带和态密度)计算案例和CASTEP[11]量子力学计算软件实践操作,帮助学生进一步巩固第一性原理计算方法、计算思路以及核心物理概念。我们以本征LiFePO4以及Mn元素掺杂LiFePO4为例,开展(掺杂)晶体结构建模、第一性原理计算优化晶体结构、非自洽计算能带和电子态密度、后处理分析。主要步骤如下:
(1) 基于Materials Project计算材料数据库,获取LiFePO4晶体结构(mp-19017),对LiFePO4晶胞进行1 × 2 × 2扩胞处理,替换超胞中的1个Fe原子为Mn从而获得LiMn0.125Fe0.875O4掺杂结构;
(2) 启动CASTEP量子力学计算模块,选择Geometry Optimization任务,选择Fine计算精度,选择GGA-PBE交换关联泛函,考虑共线自旋极化,对过渡金属Fe考虑GGA+U矫正(其中U设为3.7 eV),平面波截止动能设为500 eV,能量自洽收敛标准设置为10−5eV·atom−1,k空间采样使用Monkhorst-Pack方法和2 × 4 × 5网格,选择Ultrasoft超软赝势,其他设置参数采用软件默认值,对第一步所获得的LiFePO4及其掺杂晶体结构执行几何结构优化计算(优化晶格常数和原子坐标),待能量、力、位移均达到收敛值后,获得最优晶体结构;
(3) 针对上一步所获得的最优晶体结构,再次启动CASTEP量子力学计算模块,选择Energy单点能计算任务,选择Ultra-fine计算精度,k空间采样使用3 × 6 × 8网格,勾选能带、电子态密度等性质,其他第一性原理计算设置参数与第二步相同,再次执行计算;
(4) 启动CASTEP模块中的Analysis功能,分别执行Band structure、Density of states分析,获得能带、总电子态密度、各元素分电子态密度等数据,如图3所示。

图3 LiFePO4电子结构第一性原理计算结果
通过上述锂离子电池LiFePO4电子结构计算案例教学,让学生掌握第一性原理计算主要步骤,掌握计算材料数据库使用方法和(掺杂)晶体结构建模方法,进一步理解密度泛函理论、交换关联泛函、自旋极化、平面波截断动能、波矢k、倒易空间、布里渊区、赝势等第一性原理计算方法中的核心概念,掌握能带、电子态密度、电荷密度等电子结构性质计算方法。
3.3 锂离子电池有机电解液分子动力学模拟
电解液是锂离子电池的血液,对电解液微观分子结构的解析有助于深刻理解微观结构–宏观性质之间的构效关系,有助于提升电解液电化学性能[12]。分子动力学模拟作为一种辅助实验手段,帮助人们在原子层次理解电解液微观结构,已经被广泛地运用到电解液的研究之中[13]。我们通过在计算化学、计算材料学课程的分子动力学理论计算方法教学中引入锂离子电池有机电解液分子动力学模拟案例,进一步强化学生对分子动力学模拟方法、物理概念以及关键模拟参数的理解、掌握。我们以LiPF6: EC : EMC : DMC (1 : 10 : 10 : 10 mol%) (摩尔分数)电解液组分为例,开展电解液液体结构建模、结构优化、分子动力学模拟计算、后处理分析锂离子溶剂化结构(Li―O平均键长)和锂离子输运动力学性质(扩散系数)。主要步骤如下:
(1) 分别单独构建Li+、、EC、EMC、DMC等6个离子、分子结构,用clean功能进行初步优化;
(2) 启动Amorphous Cell蒙特卡洛计算模块,选择construction任务,导入6类离子、分子结构,其粒子数目分别为5Li+、、50EC、50EMC、50DMC,密度设为1.2 g·cm−3,选择Ultra-fine计算精度,选取COMPASS[14]力场,执行run,将溶质和溶剂分子以无定型形式随机地填充到一个周期性立方盒子中,获得电解液液态分子结构xtd轨迹文件(1帧);
(3) 启动Forcite分子(动)力学计算模块,选择Geometry Optimization任务,选择Ultra-fine计算精度,选取COMPASS力场,选取Ewald算法处理库伦静电作用,其他设置参数采用软件默认值,对第二步获得的电解液液体分子结构进行几何结构优化计算(优化原子坐标),以释放内应力;
(4) 启动Forcite分子(动)力学计算模块,选择Dynamics任务,选择Ultra-fine计算精度,选择NPT系综,模拟温度300 K,压强0.0001 GPa,粒子初始速度设置采用random (麦克斯韦-玻尔兹曼分布)方法,步长1 fs,模拟时间100 ps,选取COMPASS力场,其他设置参数采用软件默认值,对第三步几何结构优化计算获得的电解液分子结构进行分子动力学模拟,获得100 ps模拟时间内的原子轨迹、电解液密度值演化等信息,在平衡时间段内对电解液密度进行平均处理,获得平均密度信息。
(5) 基于第四步模拟所获得的平均密度,对最后一帧电解液分子结构进行体积放缩,选用NVT系综,粒子初始速度设置采用current方法,模拟时间200 ps,其他分子动力学模拟设置参数与第四步相同,再次执行分子动力学模拟;
(6) 打开第五步获得原子轨迹xtd文件,分别选择所有Li、O (EC)、O (EMC)、O (DMC),设置为对应元素或元素组的sets,启动Forcite模块中的Analysis功能,分别执行Li―O原子对的Radial distribution function (RDF)分析、Li离子的Mean square displacement (MSD)分析;
(7) 基于第六步RDF和MSD数据,通过相关公式,获得Li―O平均键长、Li―O平均配位数、Li离子扩散系数等溶剂化结构、扩散动力学等性质,如图4所示。

图4 锂离子电池有机电解液分子动力学模拟计算结果
通过上述锂离子电池有机电解液分子动力学模拟案例教学,让学生掌握分子动力学模拟主要步骤,进一步理解周期性边界条件(模拟盒子)、麦克斯韦-玻尔兹曼分布、系综、温度压强标定、分子力场等分子动力学模拟方法中的核心概念,掌握分子结构、液体(溶液)结构建模方法,掌握热力学性质、液态分子结构性质、动力学性质计算方法。
3.4 锂硫电池正极催化剂吸附多硫化物蒙特卡洛模拟
锂硫电池作为新一代高比能储能体系,却受制于正极侧硫物种反应动力学缓慢的问题,高效的硫正极催化剂可加速多硫化物转化并抑制穿梭效应,对改善锂硫电池性能具有重要作用[15]。目前,蒙特卡洛模拟等分子模拟方法已经被广泛地运用到锂硫电池正极催化剂研究[16,17]。我们在计算材料学课程的蒙特卡洛模拟理论方法教学过程中,引入Pt金属催化剂表面吸附多硫化物分子构型计算案例和Adsorption Locator蒙特卡洛模拟计算软件实践操作,帮助学生进一步巩固蒙特卡洛模拟方法、计算思路以及核心概念。我们以Pt(111)表面吸附Li2S2分子构型计算为例,开展吸附结构建模、蒙特卡洛模拟退火。主要步骤如下:
(1) 基于Materials Project计算材料数据库,获取Pt晶体结构(mp-126);
(2) 启动Cleave surface功能,对Pt体相结构进行切面操作,选择(111)晶面,原子层厚度设置为10层,真空层厚度设为15 Å (1 Å = 0.1 nm),获得Pt(111) slab表面模型,再进行5 × 5 × 1扩胞处理,得到超胞模型;
(3) Li2S2分子建模,并用DMol3量子力学计算模块进行分子结构优化(方法步骤类似案例3.1),获得Li2S2分子最优结构;
(4) 启动Adsorption Locator蒙特卡洛模拟模块,选择Simulated annealing模拟退火任务,计算精度设为Ultra-fine,吸附分子选择第三步Li2S2最优分子结构(加载量为1),退火圈数设为10,每圈退火设2000步,选取Universal[18]通用力场,选择电荷平衡算法,选取Ewald算法处理库伦静电作用,根据一个最外层Ni原子设置表面吸附区域,最大吸附距离设为5 Å,其他设置参数采用软件默认值,执行蒙特卡洛模拟退火计算,获得Pt(111)表面吸附Li2S2分子的最低能量构型,如图5所示。

图5 锂硫电池正极Pt催化剂吸附多硫化物蒙特卡洛模拟计算结果
通过上述锂硫电池金属催化剂表面吸附多硫化物分子模拟案例教学,让学生掌握蒙特卡洛模拟主要步骤,进一步理解模拟退火、分子力场等蒙特卡洛模拟方法中的核心概念,掌握表面结构、表面吸附结构建模方法。
4 实验时间安排
本实验面向高年级本科生或低年级研究生开设,实验前主讲教师指导学生安装所需软件,对量子化学计算、密度泛函理论计算、分子动力学模拟以及蒙特卡洛模拟的基本原理和流程进行简要说明,并进行主要实验流程的演示。建议本模拟实验由课上4个学时完成(具体时间安排如表1所示),课后安排学生独立撰写实验报告。
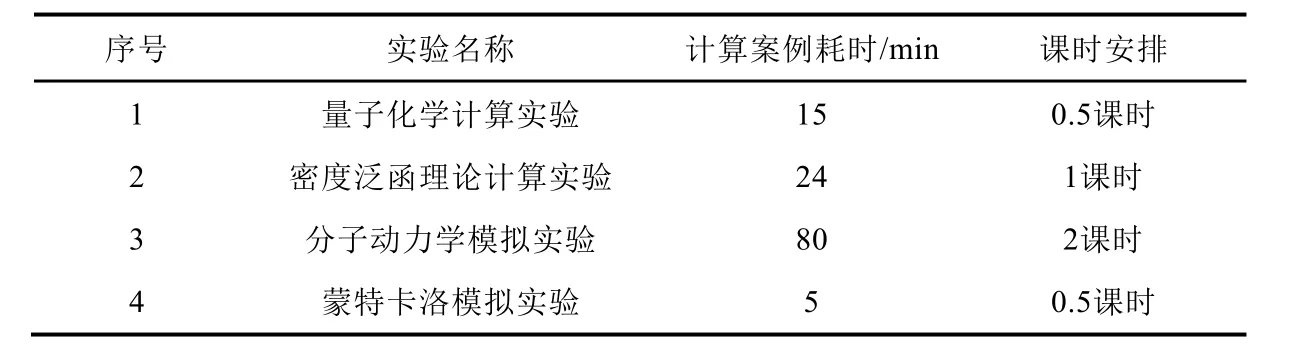
表1 模拟实验时间安排
5 总结
本论文通过介绍四个Materials Studio软件在锂电池材料计算模拟实践操作案例,展示如何采用项目式教学方法,如何将科研计算模拟案例及其软件操作有机融合到计算化学和计算材料学理论方法教学过程,让理论知识概念落脚于具体的计算案例和计算操作,使教学内容从抽象到直观。同时,让学生领略到计算模拟对现代化学、材料研究的巨大推动作用,培养学生科学素养和科学思维,实现科研反哺高质量教学。在理论方法教学中使用Materials Studio科研工作软件,不仅增强教学内容趣味性、提高课堂教学效率,更为重要的是提升学生科研探索精神和培养科研思维能力,实现教学支撑高水平科研。除了上述四个实例之外,在材料结构表征(振动频率计算,如红外光谱与拉曼光谱等)、化学反应热力学定量计算(如焓变、热力学相图等),以及化学反应或离子扩散动力学计算(如过渡态能垒、锂离子迁移能垒)等诸多方面,Materials Studio等科研工作软件皆可大显身手,用以辅助理论计算方法教学。笔者将持之不断地探索Materials Studio等科研工作软件在未来的理论计算方法教学过程中的融合应用。
