辨析基础有机化学教材中的争议问题:乙烷构象稳定性的来源
2024-03-04凌皓博赵佩鈜俞寿云
凌皓博,赵佩鈜,俞寿云
南京大学化学化工学院, 南京 210023
烷烃的构象及其稳定性是基础有机化学中重要的知识点,其中乙烷的交叉式与重叠式构象是其中最基础的部分。乙烷的交叉式构象比重叠式构象稳定约13 kJ·mol−1(图1),这一事实在国内外的主流基础有机化学的教材中没有争议。但是我们发现,对于“乙烷交叉式构象比重叠式稳定”这一事实,国内外不同教材给出的解释大不相同,我们将其中代表性的解释列在表1中以供参阅[1–10]。
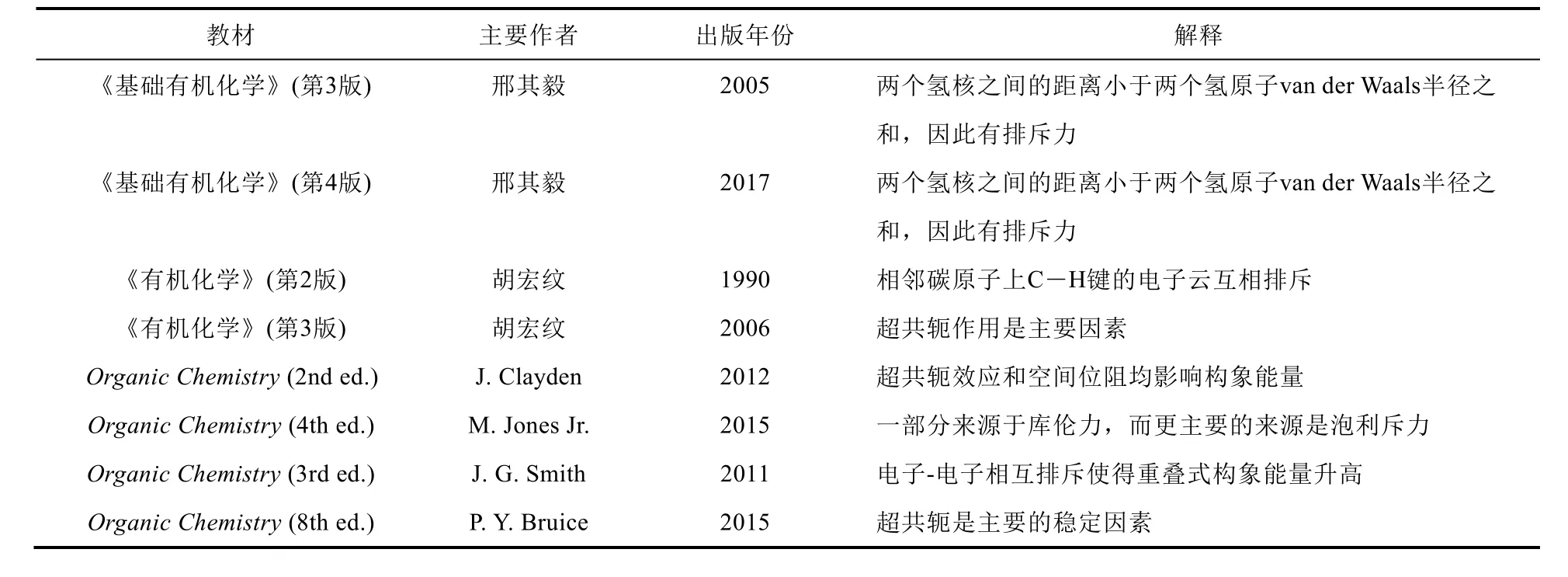
表1 代表性教材对乙烷交叉式构象比重叠式构象稳定的解释
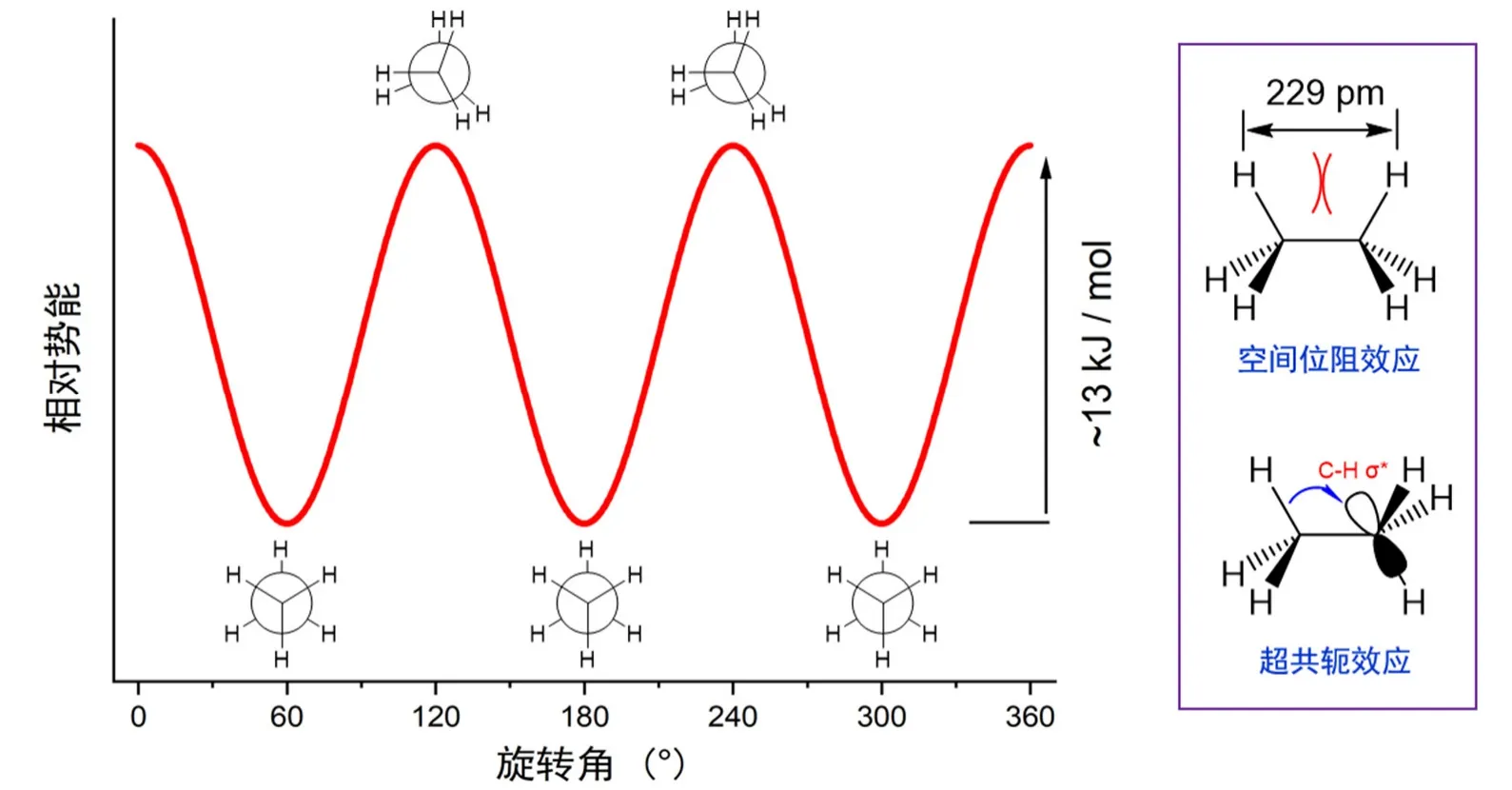
图1 乙烷构象的势能图及其解释
从表1可以看出,国内不同的教材对交叉式构象比重叠式稳定的解释各不相同。比如,邢其毅等著的《基础有机化学》(第3版)认为氢核间距(229 pm)小于两个氢原子范德华半径(120 pm)之和,从而导致重叠式能量升高[1],第4版也未对其观点进行更新[2]。胡宏纹等著的《有机化学》(第2版)认为是C―H键电子云相互排斥使得能量升高[3]。2001年,Pophristic和Goodman在Nature发表论文,通过理论计算认为超共轭效应是乙烷交叉式构象稳定的主要因素[11],周公度在《大学化学》上及时地对这篇Nature论文进行介绍和评述[12]。胡宏纹等著的《有机化学》(第3版)[4]及之后的版次[5,6]均采纳了“超共轭效应是乙烷交叉式构象稳定的主要因素”这一观点。国外的主流教材同样众说纷纭,Clayden等人认为空间位阻效应和超共轭效应均对乙烷构象的稳定性有影响且没有说明哪种因素占主导[7]。而其他的几本国外基础有机化学教材虽然是在2010年后出版的[8–10],但观点也不统一,这很不利于初学者的学习。
为了辨析乙烷构象稳定性的来源问题,我们调研了近百年来对该问题研究的代表性文献,并将文献中的研究方法与结果进行对比分析,同时借鉴了现代计算化学的最新成果,旨在阐明该争议的形成原因并介绍目前的研究进展情况,以便后续教材编写修订时的参考和初学者的理解掌握。
1 文献调研及分析
自1927年W. Heitler和F. London提出价键理论以及1932年Eyring等人[13]用氢分子之间的势能曲线估算了乙烷重叠式构象和交叉式构象的能量差以来,乙烷交叉式构象为何比重叠构象稳定以及乙烷构象转变能垒来源的问题一直是化学家们研究的重要科学问题之一,也是争论的焦点问题之一。随着理论、计算方法和计算工具的发展,科学家对这一问题的解释总体上呈现由简单到复杂、由粗糙到精确的变化趋势。
已有的研究中,对乙烷构象稳定性差异及构象转变能垒来源主要有三种因素:库伦力(Coulombic Force)、泡利斥力(Pauli Repulsion,也称为交换排斥,Exchange Repulsion)和超共轭效应(Hyperconjugation)。另外,范德华力(van der Waals Force)也被认为是一种可能的因素。库仑力是指静止电荷之间遵循库仑定律的相互作用;泡利斥力是指当两个原子接近时,两原子上相同自旋的电子由于泡利不相容原理的作用,导致原子间的电子密度减小,电子云对核间排斥的屏蔽效应减弱,使两原子的核间斥力增加,总体系能量升高;超共轭效应是指交叉式乙烷的一个C―H键中的电子离域到另一碳原子C―H键的反键轨道上造成能量降低的现象;范德华力是指分子间或分子内各原子和原子团的永久偶极、诱导偶极和瞬时偶极之间的相互作用。其中,库仑力和泡利斥力的影响统称“空间斥力”或“位阻因素”,科学家的主要争论点就在于乙烷构象稳定性差异及构象转变能垒来源于空间位阻还是超共轭效应,以下将这两种观点分别称为“位阻学说”和“超共轭学说”。
乙烷构象稳定性问题的研究历程按照研究中使用的理论和计算方法,大致可以分为三个阶段,分别为前量子化学时期(约1960年以前)、早期量子化学时期(约1960–2000年)和当代量子化学时期(约2000年以后)。由第一阶段进入第二阶段的主要标志是现代量子化学的基础——Hartree-Fock近似和一般精度的量子化学计算的广泛使用;随着高精度量子化学计算方法的逐步发展,由第二阶段进入第三阶段,主要标志是2001年Pophristic和Goodman在Nature杂志发表的一篇论文[11],他们强调“超共轭效应是乙烷构象稳定性差异及构象转变能垒的来源”,这一观点引发激烈争论。表2总结了各个阶段的一些代表性研究论文,反映了对该问题研究的脉络和争议情况[13–33]。
1.1 前量子化学时期(约1960年以前)
这一时期,现代量子化学的计算方法尚未建立,研究者用一些较为简单和粗糙的方法来解释乙烷构象稳定性差异,常用的理论有价键理论和原子轨道线性组合分子轨道法(LCAO-MO),得出的结论也多有争议。例如,1932年Eyring用价键理论计算氢分子之间的各种相互作用曲线,接着用这来估算乙烷不同甲基的氢之间的相互作用,认为位阻因素能解释乙烷C―C键的转动能垒[13]。1939年,Eyring等用同样方法计算的结果却不支持这一解释[14]。在价键理论框架内,对相互作用能的不同数学处理方式也对结论有一定影响。例如,1949年,Dean用幂级数近似相互作用能,支持了位阻学说[17],而1951年,Oosterhoff用三角级数近似却不支持位阻学说[18]。
值得注意的是,在这个阶段,尽管位阻学说是主流观点,超共轭学说已经开始萌芽。例如,分子轨道理论的提出者之一Mulliken在1939年就提出乙烷中可能存在“超共轭效应”[15]。他类比含有C≡N三键的氰基和含有三个C―H键的甲基,认为氰(NC―CN)和乙烷分子内两基团之间的相互作用有一定共性,把它们分别称为“共轭”和“超共轭”,并在1941年用早期的分子轨道理论进一步阐述了这个观点[16]。甚至价键理论的分析也间接承认了电子的某种“离域性”。例如,1958年Eyring等参考了1939年Mulliken的观点,提出乙烷中有所谓“H―C―C―H离域键”,并用以解释乙烷构象稳定性差异[19]。
尽管理论和计算方法的局限性以及计算的精度限制了这一时期对乙烷构象稳定性差异及构象转变能垒来源的讨论,它们仍然为更深入的讨论和更精确的理论计算奠定了基础。
1.2 早期量子化学时期(约1960–2000年)
这一时期,计算方法和计算工具有所进步,求解多电子薛定谔方程的基本方法——Hartree-Fock自洽场近似等计算方法的产生和计算机技术的发展提高了量子化学理论分析的精度。20世纪60年代的Hartree-Fock近似[20]和Hellmann-Feynman近似[21]计算都支持位阻学说。然而,这些计算中的一些假设受到了质疑。例如,Honegger在1989年指出重叠式乙烷C―C键长大于交叉式乙烷,使得前者两个甲基的氢之间的库伦斥力反而更小,甚至使得重叠式更加稳定[26]。过去的一些研究忽略了这一差别,得出的结论存在不足。
1970年,Lowe用分子轨道理论进行定性分析,首次提出乙烷一个C―H键的电子向另一个C―H键的反键轨道离域,确立了现代版本的超共轭学说[22]。次年,England和Gordon用间略微分重叠(INDO)近似计算支持了这一观点[23]。从此,位阻和超共轭两大学说相互争论和补充的局面形成。Pople[24]、Bandenhoop[27]等讨论了结合位阻和超共轭两种学说来解释乙烷构象稳定性差异的可能性,Stevens和Karplus[25]则对比分析多种计算方法试图捍卫位阻学说。
1.3 现代量子化学时期(约2000年以后)
2001年,Pophristic和Goodman将影响乙烷构象稳定的因素分为三部分[11]:泡利斥力、库伦力(这两个合称为位阻因素)和超共轭因素,然后采用将位阻因素和超共轭因素分别剔除,再用自然分子轨道法进行模拟计算的方法,发现了一个惊人的结果(图2):如果去除位阻排斥作用,乙烷构象将仍然倾向于交叉式,而如果去除超共轭作用,则乙烷的构象会偏向于重叠式,与事实完全相反。他们认为这是因为在交叉式向重叠式的旋转过程中C―C键会伸长,构象变化已经不是单纯的旋转,还包含了碳骨架的扩张,从而消解了本应出现的位阻斥力,让位阻不再是影响乙烷构象偏好的重要因素。
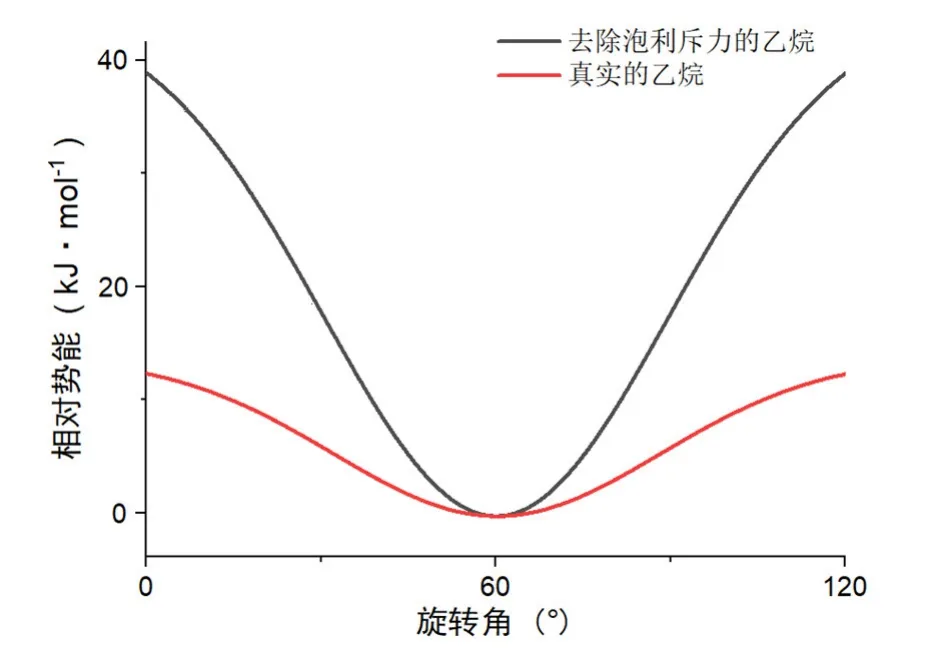
图2 真实乙烷和去除泡利斥力作用的乙烷势能图
但是文章中有些问题没有澄清:1) 库伦力、泡利斥力和超共轭效应对能量的影响是否相互独立?“分别剔除”这一方法得到的计算结果能否有说服力?2) C―C键伸长所带来的能量变化是否需要考虑?这些不完善的地方立刻被位阻一派抓住予以反击。
2003年,Baerends采用将两个甲基自由基拼接构造C―C键的方式,否定了Goodman等人文章中“位阻因素偏向重叠式构象”和“位阻因素对构象没有影响”等结论[28]。他们将C―C键长和甲基构型分别固定为重叠式和交叉式构象所对应的情况,然后进行旋转,得到如图3的能量曲线:在构象固定时,重叠式构象无论如何位阻都会更大,而在实际情况中,C―C键的伸长使得斥力减小,让人误认为“重叠式的位阻排斥更小”,实际上正是由于位阻排斥变大,才会迫使C―C键伸长来减小斥力,与经典解释相符。不过让人疑惑的是,从能量图中看,交叉式构象的能量应该更高,仿佛与结论自相矛盾。对此他们认为原因在于能量优化函数,即两个自由基成键生成真实分子的过程中会发生“整体性能量优化”使得能量会发生相对变化,其在图3中并未体现出来[34]。然后,他们将不同能量项都化成C―C键长度的函数,通过该计算得出的结论是,无论在什么情况下,泡利斥力都牢牢的占据主导位置,即它的变化值远大于其他因素所造成的能量差值。根据此文献的计算,超共轭所带来的交叉式构象的能量减少只有1.6 kJ·mol−1,甚至还会被库伦力所带来的1.2 kJ·mol−1的能量升高所抵消。

图3 三种不同情况下随旋转角改变的泡利斥力能图
2005年Mo[29]和2007年Gao[30]两个研究团队使用从头计算的价键理论,均认同了位阻因素是交叉式乙烷构象更为稳定的主导因素。但是,价键理论本身同样存在不足,就像Baerends所指出的那样,通过价键理论构造完整分子的过程中,有些能量变化实际上是被忽略掉的。2016年, Wierzbicki使用CC/MBPT(耦合簇/多参考微扰理论)计算,再次支持超共轭一派的观点,认为位阻效应对于13 kJ·mol−1的转动能垒几乎毫无贡献[31]。2022年Jenkins使用QTAIM理论进行模拟计算,认为先前价键理论中的泡利斥力被高估了,他们所模拟出的斥力只有原先的1/2[32]。2023年,Chaquin使用动态轨道力(Dynamic Orbital Forces)的方法进行模拟,发现重叠式中的超共轭作用并不比交叉式的超共轭要弱很多,泡利斥力仍是乙烷构象的主导因素[33]。
即使运用了现代的计算方法,“位阻学说”和“超共轭学说”双方都仍然没能拿出足够有说服力的证据来结束这场争议,两派的争论一直持续到今天,还将继续下去。
2 结论与建议
2.1 结论
对于乙烷交叉式构象比重叠式构象稳定的原因,超共轭效应和位阻效应谁占主导的问题至今仍然未能达成共识,不过位阻效应和超共轭效应均影响乙烷构象稳定性这一事实已经十分清晰。实际上,每一种计算方法都需要先通过对体系进行不同形式和精度的近似后才能进行模拟计算,而根据选取的计算方法不同,在这个问题上得到的结果也会不同[33]。整体而言,各个研究者所选取的主要方法可以分为两大类:自然分子轨道法(NBO)和现代价键理论(VB)。使用自然分子轨道法时,需要通过撤除空的轨道来计算超共轭效应和全充满的σC―H轨道之间排斥效应的相对权重,Goodman、Weinhold都采用这个方法并得出超共轭占据主导的结论。但是后续文献证明这一影响被高估了,在加入超共轭因素的现代价键理论计算中,位阻效应仍然占据主导,与前面结论正好相反。
我们暂时无法得知哪种近似方法与实际情况相比误差更小,但两种方法及观点都有其价值,至于哪一个效应占主导,期待后续研究者能发展出更加精确的计算方法来彻底解决这个争议。
2.2 建议
我们建议教材编写者在编写乙烷构象稳定性的章节时,将两种因素均进行说明,即乙烷构象偏向于交叉式是位阻效应(主要来源于泡利斥力而非库伦力)和超共轭效应共同作用的结果。在课程讲解的过程中引导学生阅读原始文献,厘清这一问题的研究脉络,自己得出结论。
