抗GQ1b、GT1a、Sulfatide抗体阳性的类重症肌无力Miller-Fisher综合征1例报告
2024-02-29王天舒张旭王立波刘松岩
王天舒, 张旭, 王立波, 刘松岩
1 病例资料
患者,女,48岁。因“双侧上睑下垂、视物模糊5 d”入院。患者入院前5 d无明显诱因出现双眼睑下垂、视物模糊,自诉晨轻暮重,劳累后症状加重,休息后症状减轻,同时伴头晕及四肢无力麻木,未经特殊治疗,症状持续进展。曾就诊于当地医院,行眼底检查未见明显异常,排除青光眼、葡萄膜炎等眼科疾病。否认既往感染史及手术史,近期无眼部外伤史、散瞳史、药物服用史、有毒气体吸入史及肉毒素注射史。入院时查体:神志清楚,言语流利,左侧眼睑下垂至角膜1/5处,右侧眼睑下垂至角膜1/4处,双侧眼球固定,复视(+),无眼震,双侧瞳孔形状呈梅花状,双侧瞳孔扩大至5~6 mm(见图1),双眼直接、间接对光反射迟钝(见图2),四肢肌力Ⅴ级,感觉查体未见明显异常,双上肢腱反射减弱,双下肢腱反射正常,右侧指鼻试验欠稳准,双侧跟膝胫实验稳准,双侧病理征阴性;新斯的明实验前定量MG评分(quantitative MG score,QMGS)8分,实验后改善最显著时QMGS为4分,相对评分为50%,可疑阳性,疲劳试验可疑阳性。综上,该患以眼肌麻痹、共济失调及瞳孔形状和瞳孔对光反射异常为主要临床表现,病情具有病态疲劳波动性特点,需要鉴别重症肌无力、Miller-Fisher综合征和Adie综合征。完善血液、尿液、脑脊液常规生化、影像检查、肌电图、肌无力抗体及自身免疫性周围神经抗体检查(见表1),结果显示脑脊液蛋白细胞分离,抗GQ1b、GT1a、Sulfatide抗体阳性,根据2019年中国吉兰-巴雷综合征诊治指南,本例患者存在眼肌麻痹、共济失调、双上肢腱反射减弱、脑脊液蛋白细胞分离特征及血清抗GQ1b抗体阳性,临床诊断为Miller-Fisher综合征,给予激素冲击(甲泼尼龙0.5 g/d,2 d后减量为甲泼尼龙0.25 g/d,持续3 d)、营养神经等对症治疗后5 d症状好转,双眼睑于角膜上缘,双眼球各向活动自如,双侧瞳孔等大同圆,直径约3 mm(见图3),直接、间接对光反射灵敏(见图4),出院后口服激素治疗并逐渐减量,1月后停药,停药后随访无明显不适。

表1 辅助检查结果

图1 治疗前瞳孔形状及大小

图2 治疗前对光反射示意图
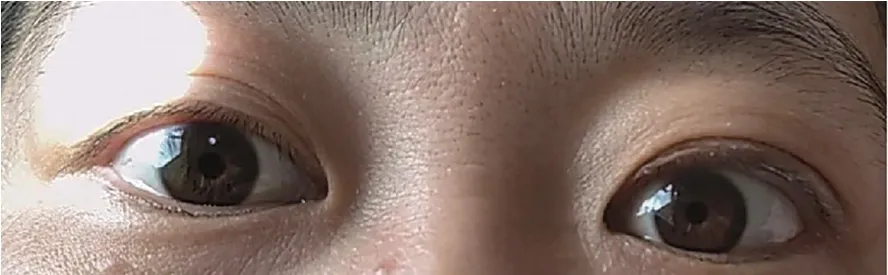
图3 治疗后瞳孔形状及大小
图4 治疗后对光反射示意图
2 讨论
MFS是吉兰-巴雷综合征的一种变异型,以眼肌麻痹、共济失调和腱反射消失为主要临床特点[1],常伴抗GQ1b抗体阳性。本例患者具备眼肌麻痹和共济失调特点,同时还出现抗Sulfatide、GT1a血清抗体阳性,眼内肌麻痹以及症状波动现象。下面将针对这3点进行讨论。
自1992抗GQ1b抗体就被用作MFS的诊断标志物,GQ1b属于神经节苷脂的一种,研究显示GQ1b分布在动眼神经、外展神经、滑车神经、舌咽神经、迷走神经的髓外部分和神经肌肉接头,以及纺锤体的Ia类传入纤维和背根神经节的一些大神经元[2],故MFS三联征的可表现为眼肌麻痹、腱反射减弱和共济失调,抗GQ1b抗体也可见于急性眼肌麻痹、急性共济失调、Bickerstaff′s脑干脑炎和咽-颈-臂(pharyngeal-cervical-brachial,PCB)变异型,临床表现的差异性可能与抗体结合部位以及多重抗体反应性改变相关[3]。本例患者除GQ1b抗体阳性,Sulfatide、GT1a血清抗体均为阳性,Sulfatide作为硫脂大部分存在于中枢和外周的髓鞘细胞中[4],Sulfatide抗体阳性可见于多种周围神经病变,包括急性脱髓鞘性多发性神经根炎[5]、慢性脱髓鞘性多发性神经根炎[6]、脑血管病或其他疾病[7],目前关于Sulfatide抗体对于临床疾病的诊断价值仍需进一步研究,不认为抗Sulfatide抗体为该患者责任抗体。PCB最具诊断意义的抗体为抗GT1a抗体,其次为抗GQ1b抗体,GT1a在支配口咽部及颈臂肌肉的神经上集中表达,故PCB表现为快速进展的咽喉、颈部肌肉及双上肢无力和腱反射减弱[8],GT1a和GQ1b由同一前体组成[9],二者具有部分相同结构,抗体间可发生交叉反应[3],当出现两种及以上变异型临床表现时可诊断为重叠综合征[10],但本例患者并无PCB表现,认为抗GQ1b抗体为该患者的责任抗体,故仅诊断为MFS。
本例患者特殊表现出眼内肌麻痹:瞳孔扩大,瞳孔形状改变及对光反射迟钝,需要与 Adie综合征鉴别。Adie综合征的特征是瞳孔散大,直、间接对光反射减弱或消失,瞳孔收缩障碍和调节反射异常,合并下肢腱反射消失或发汗异常,最具特征性的体征为光-近分离,其病因是不明原因副交感睫状神经节受损导致,病程具有持续不缓解特点。本病例脑脊液出现“蛋白细胞分离”现象以及血清中检出抗GQ1b、Sulfatide、GT1a抗体,排除Adie综合征,确诊为MFS。中国吉兰-巴雷综合征诊治指南提到极少数患者可出现眼内肌受累情况,少量病例报道提及眼内肌受累的MFS[11,12],结合MFS和Adie综合征的发病机制,考虑是自身抗体攻击与动眼神经一同走行的副交感神经,导致瞳孔散大,其中损伤较轻副交感纤维使得瞳孔扩约肌节段性收缩,故瞳孔边缘呈梅花状。
除此之外,本例患者症状还表现出类重症肌无力波动性特点,MFS 和MG均是自身免疫性疾病,可产生针对周围神经和神经肌肉接头的自身抗体,本例患者尽管新斯的明实验可疑阳性,但血清肌无力抗体未检出,重复电刺激未见波幅衰减,而且脑脊液中出现典型的“蛋白-细胞分离”现象,不支持MG诊断,考虑是由抗GQ1b抗体导致患者出现类肌无力症状,这一猜想在动物模型中得到证实[13],含有神经节苷脂的脂质参与了电压依赖型钙离子通道(voltage dependent calcium channel,VDCC)的聚集和囊泡释放区的形成,血清中抗GQ1b抗体与突触前膜的神经节苷脂结合后[2,14],一方面影响囊泡释放,另一方面在补体级联作用下形成膜攻击复合体[15],致使钙离子迅速进入胞内导致细胞胞吐障碍[16],最终使突触间隙的乙酰胆碱浓度降低,出现类似重症肌无力的临床表现,尽管本例患者肌电图未见明显异常,但有报道提到MFS患者肌电图出现类似重症肌无力肌电图特征[10],也有报道中提到MFS患者和MFS小鼠模型在重复电刺激(repetitive neural stimulation,RNS)后出现增量反应[17],该反应与Lambert-Eaton肌无力综合征(Lambert-Eaton myasthenic syndrome,LEMS)的肌电图特征相似[18],故抗GQ1b抗体与突触的结合部位仍需进一步探索。
MFS是较为罕见的一种疾病,临床上应与脑干卒中、糖尿病性眼肌麻痹、视神经脊髓炎、多发性硬化和重症肌无力等相鉴别,当病例合并瞳孔改变时应与青光眼、虹膜炎、阿罗瞳孔、Adie综合征等鉴别。
伦理学声明:本研究经由吉林大学中日联谊医院伦理委员会审批(批号:2023112701),并免除了获得书面知情同意的要求。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:王天舒、张旭负责文献查阅收集、拟定写作思路及撰写文章;王立波、刘松岩负责论文审阅及修改。
