图卷积神经网络在晶体材料研发中的应用进展
2024-02-27劳思思田子奇
劳思思,田子奇
1.宁波大学,浙江 宁波 315211
2.中国科学院宁波材料技术与工程研究所,浙江宁波 315201
引 言
深度学习技术近年来深受研究人员的关注,强大的功能使其在计算机视觉、自然语言处理等领域取得了突破性进展。与传统的机器学习方法相比,深度学习建立了人工神经网络,可以对更深层的特征进行提取与识别,使机器能学习到更加抽象的概念,大大增强了其表征学习的能力。其中,卷积神经网络(Convolutional Neural Network, CNN)在图像处理和语音识别等方向上体现出了尤其优异的性能[1-3]。
已有的多数研究往往针对欧式结构的数据展开,它们具有固定尺寸和维度,例如图像、文本等。对于欧式空间的数据结构,如若将图像的每一个像素点视为一个节点,相邻节点之间用一条边表示,那么每个节点具有固定数量的邻居节点,卷积操作可利用固定大小的卷积核来学习图像的特征。但现实应用中存在许多数据关系,如社交网络、交通流量、分子结构等图结构数据,均属于非欧式空间结构。在数学表示上,这些数据的特征矩阵维度并不统一。例如,对于材料和化学研究中关注的分子、晶体体系,如若将微观结构中的每一个原子视为一个节点,相邻原子之间连接的键用一条边来表示,则每个节点所具有的邻居节点的数量不固定,传统的CNN难以直接对其进行卷积和池化。如何将CNN等深度学习的算法应用于此类结构的数据是具有挑战性的课题。2014 年,Estrach 等人[4]提出图卷积神经网络(Graph Convolutional Network,GCN)概念,通过对节点之间的特征进行聚合,得到图中每个节点规整的表达形式,以其作为GCN 的输入。此时GCN 便能利用不同尺度的信息,得到更深层次的特征。之后,不同架构的GCN相继被构造出来,用于分子、晶体体系的构效关系研究和预测。
本文在第1 节中归纳和梳理了近年来GCN的发展过程和研究现状,对其主要方法、应用任务及优缺点进行简要分析;在第2 节中给出GCN在晶体材料的结构与性能研究领域的具体应用范例;在第3 节总结了目前GCN 模型的缺陷和不足,并对未来其发展方向和趋势进行了展望。
1 图卷积神经网络模型
图卷积神经网络是针对图结构进行深度学习的方法,其算法过程如图1 所示。图(graph)的概念源自图论,是一种由若干个节点(node)及连接两个节点的边(edge)构成的图形,用来描述不同结点之间的关系。每个节点均具有各自的特征,并同时受到近邻节点以及更远节点的影响而改变直至最终平衡,关系越密切的近邻节点影响越大。在操作中,图中每个节点自身特征经过非线性变换后传递给邻居节点,然后每个节点将邻居节点的特征与自身特征进行融合,再将融合后的特征进行非线性变换,通过重复该过程来加强模型的表达能力。目前主流的图卷积神经网络方法主要有两类:基于频域的方法和基于空间域的方法。
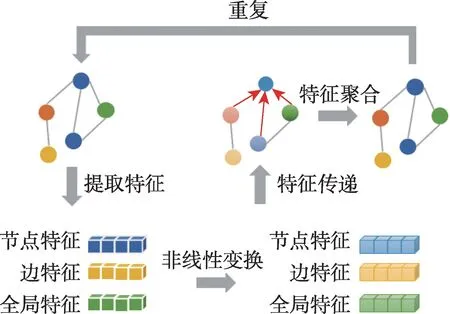
图1 图卷积网络算法过程Fig.1 Graph convolutional network algorithm process
基于频域的GCN[4]将处于节点域的输入信号转化为谱域上的信号后,利用卷积定理实现图卷积操作,再将其转化回节点域,完成对节点间的信息聚合,最后通过非线性的激活函数将其堆叠为多层的神经网络。近年为了降低模型的复杂度并解决模型非局部化问题,大量研究者提出了不同的改进方案,例如Defferrard等人[5]提出ChebyNet,使用多项式滤波器,选择中心节点及其一阶邻居输入模型;Xu等人[6]提出图小波神经网络(GWNN),用小波变换的基取代傅里叶变换的基;Zhang 等人[7]则在网络中运用威斯费勒-莱曼算法(Weisfeiler-Lehman)[8]对节点特征进行聚类。总体而言,基于频域的GCN 方法有完备的理论基础,但不及基于空间域的GCN方法直观,目前在材料研究领域运用较少。
与之相比,基于空间域的图卷积网络方法从节点的空间关系出发对图结构进行探究,通过考察中心节点与邻居节点的关联直接进行卷积操作,降低了模型的复杂度,提高了运行效率。材料和化学领域的研究体系——如晶体、分子等,往往可以表示为通过节点和边表示的图结构,因此基于空间域的GCN 是目前材料设计与预测研究中的主流方法[9]。基于空间的图卷积网络存在3 个重要问题:(1)中心节点的确定;(2)邻居节点的确定以及定序;(3)邻居节点特征的聚合。经典的面向图像的卷积神经网络框架是通过先确定节点的领域,再进行领域内的节点定序,最后实现参数共享。然而图结构中节点领域内节点数量不固定,领域内节点难定序,导致了参数共享困难。研究者选择通过某种邻近度度量来确定邻居节点,以及进行领域内定序[10],或是直接选择随机采样和重要性采样的方法,不再对节点定序,直接将邻居节点的信息通过聚合的方式来将信息联合更新,产生下一层的节点信号[11-12]。为此Kearnes 等人[13]在利用GCN 虚拟筛选小分子的过程中,采用由高斯核实现的模糊直方图来对特征进行提取,进一步减少了在特征表示学习过程中造成的信息损失。Faber 等人[14]通过简化Kearnes 等人的模型并利用原子间的欧氏距离对卷积过程中的特征向量进行运算,取得了更好的效果;Schütt 等人[15]提出SchNet 架构,通过输入原子序数和原子坐标利用连续滤波器卷积层,来建模原子系统,进一步精确预测分子和材料在化学空间中的性质;Gilmer等人[16]提出图消息传递神经网络(MPNN)框架,归纳了多种图卷积神经网络的算法流程(如图2 所示),总结出通用的GCN 过程分三步:消息传递、信息聚合以及节点更新。MPNN 将卷积算子推广到了非欧式空间的数据,实现了图结构与神经网络的连接。在MPNN 框架的启发下,Yang 等人[17]提出了定向消息传递网络(DMPNN),将原始MPNN中对节点信息的聚合和更新改变为对边信息的聚合和更新,通过避免消息传递过程中不必要的循环,减小引入噪声的可能性。但DMPNN仍忽略了原子键与节点之间的信息传递,导致两者之间无法实现消息共享。为了解决这一问题,Song 等人[18]提出了一种通信消息传递网络(CMPNN),加强了原子键与节点之间的信息交互过程,使得两者信息在模型训练过程中均得到更新。

图2 图消息传递神经网络框架流程Fig.2 Graph message-passing neural network framework flow
特别地,面向晶体类材料的研究,Xie等人[19]提出晶体图卷积网络(Crystal Graph Convolutional Neural Network, CGCNN)。如图3 所示,将晶体结构表示为一个无向多重图,节点表示原子,边表示原子之间的键。节点i的属性用特征向量vi来表示,第i个原子与第j个原子之间的第k个键用u(i,j)k来表示。晶体图与其他图结构相比不同之处在于:因为晶体具有周期性,所以它允许在同一对节点之间有多条边。该框架可以直接从晶体中原子的连接性学习材料属性并提供了具有通用性和可解释性的晶体材料的表示。Chen等人[20]意识到目前的图卷积网络模型在对材料领域进行状态预测(如自由能)时缺乏对全局状态(如温度)的描述,在节点向量与边向量的更新过程中联合了全局状态,改进CGCNN 模型为通用材料图网络(Materials Graph Network, MEGNet)模型,架构如图4 所示。模型的元素嵌入层中编码了周期性的化学趋势,利用迁移学习预测带隙和弹性模量。原始的CGCNN无论所代表的晶体结构如何,固定每个原子具有12 个邻居节点,因而可能引入一些掩盖了重要信息的节点。Park 等人[21]根据Voronoi 图来确定节点的领域,考虑了相邻原子的三体关联性,并设置了一个新的卷积函数来对边向量进行更新。因为晶体结构中具有的金属键和离子键,原子之间的连通性较高而上述的预测模型未有效地捕获原子和键之间的消息相互作用。因此,Chen等人[22]基于CMPNN框架开发了晶体网络(CrystalNet),在预测材料特性时的性能比其他图神经网络模型高29.2%。

图3 CGCNN晶体结构表示Fig.3 CGCNN crystal structure representation
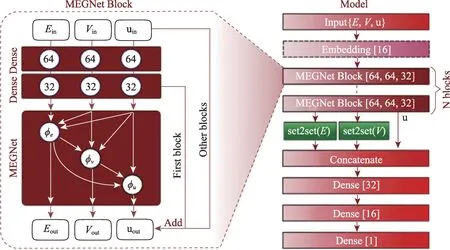
图4 MEGNet流程框架Fig.4 MEGNet process framework
为了让图卷积网络自动进行参数化的卷积核学习,Veličković等人[23]提出在图卷积网络中引入注意力机制,让模型自动学习节点与邻居节点之间的重要性。在材料科学中,Coley等人[24]首次提出了将基于全局注意力机制的图卷积神经网络用于化学反应性的预测。首先模型学习识别最可能发生连通性变化的活性位点,然后根据化学价规则对相互作用可能产生的产物进行枚举并对其进行排名。在这个模型中,每个原子的全局注意力系数是通过该原子对所有其他物理上可能匹配的反应概率来计算的。Louis 等人[25]提出的基于全局注意力机制的图卷积神经网络(Global Attention Mechanism with Graph Neural Network,GATGNN)中原子的注意力系数的计算是根据:(1)原子在图中位置的重要性;(2)原子在材料结晶过程中贡献的能量。GATGNN体系结构如图5所示,每个模型由5或7 个增强图注意力层(Augmented Graph-Attention,AGAT)组成,共有64 个神经元。模型在提取节点特征后,先设置全局注意力层来学习参数,然后再对特征向量进行全局池化。将晶体的特征向量进行加权求和后输入到两个隐藏的线性层,最后再连接一个全连接层用来输出预测的晶体属性。表1 对图卷积网络方法的优缺点,应用任务以及时间复杂度或相关函数进行了简单汇总。
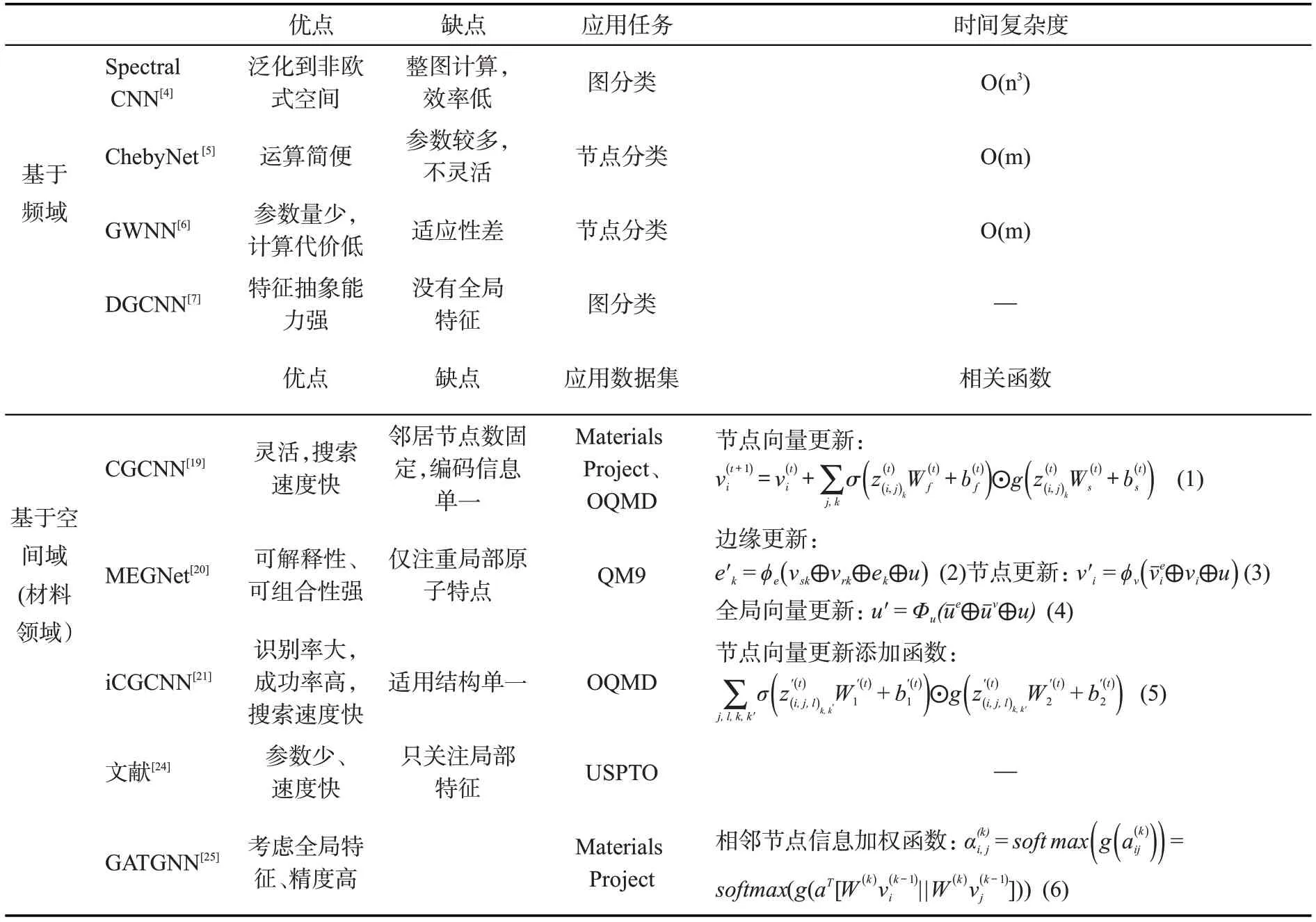
表1 各个图卷积网络模型比较Table 1 Comparison of various graph convolutional network models
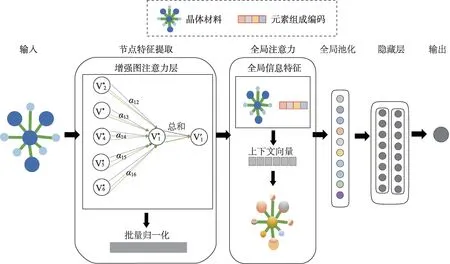
图5 GATGNN体系结构Fig.5 GATGNN architecture
图卷积神经网络为处理非欧式空间的图结构数据开辟了新的途径,通过与迁移学习、注意力机制等人工智能技术结合,使网络能够准确快速地提取图结构中的特征信息。材料科学研究的结构体系中原子与原子之间的键,天然形成了图结构,为图卷积网络在分子、晶体体系中的应用奠定了基础。下文对目前图卷积神经网络在晶体材料中的应用进行简要介绍。
2 图卷积神经网络在晶体材料中的应用研究
2.1 稳定结构的预测
材料的结构决定了材料的性质,预测稳定结构是材料探索的基本问题。图卷积神经网络模型可以基于晶体材料的各种特征进行预测,寻找出新的稳定的晶体结构。方法可分为正向设计与逆向设计两大类:正向设计是利用人的先验知识,替换已知稳定结构中的元素,在得到数据集后,利用图卷积网络模型对相关性质进行预测,以找到新的材料体系;逆向设计则是通过一定算法,从头构建符合目标属性的材料结构。
采用正向设计的思路,Xie 等人[19]对于形式为ABX3的钙钛矿晶体结构(A、B为任意非放射性金属,X 为O、N、S 和F 中的一个或几个元素)构建结构数据库,利用CGCNN来预测数据库中钙钛矿形成能,运用组合搜索寻找稳定的钙钛矿,在18,928个结构中找到228个存在合成可能性的钙钛矿型材料。Venturi 等人[26]将CGCNN扩展到具有平面对称性的二维过渡金属碳化物和碳氮化物(2D MXenes),使用了100 个CGCNN模型的集合,每个模型在计算材料数据库(Computational 2D Materials Database,C2DB)中随机选取70%的数据作为数据集进行训练,通过额外增加校准和锐度来衡量模型的性能。利用训练好的模型对HOIP[27]、Castelli[28]、aNANt[29]3个数据库的材料进行筛选,该模型预测的生成焓误差均低于0.1 eV atom-1,与DFT计算误差精度相当。ThCr2Si2型结构是自然界中最常见的晶体结构之一,Park 等人[21]用52 个非稀土金属元素依次替换ThCr2Si2结构中的相应元素,得到132,600个ThCr2Si2型化合物,通过CGCNN方法筛选出了97 个稳定的结构。由于原始的CGCNN不考虑结构弛豫的问题,可能会在预测过程中造成误差,Juhwan Noh 等人[30]利用材料工程数据库(Material Project, MP)中已存在的Mg-Mn-O 三元化合物作为模板,将各种元素替换到已知的晶体模板中,生成7,000 多个虚拟的晶体结构。通过改变CGCNN 原始网络中的激活函数,并在网络中增加正则化操作,预测晶体形成能来搜索稳定结构。相比原始的CGCNN模型预测未经结构优化的晶体结构形成能,预测准确率提高了10%~15%。上述对于晶体材料的设计方法,只通过考虑热力学形成能来推断材料的合成性,判定标准过于简单。为此,Jang等人[31]提出一种基于正样本和无标签学习(PU learing)的优化方法,运用图卷积神经网络作为分类器输出合成分数,来量化晶体材料的合成概率。基于2014 年的ICSD 和MP 数据库训练模型,其真阳率为86.2%。Gu等人[32]将实验制备的943 个钙钛矿晶体结构和11,964 个虚拟的钙钛矿晶体结构作为数据集,把图卷积网络与正样本和无标签学习(PUL)[33]、领域特定学习(DSL)[34]和迁移学习(TL)[35]相融合,运用组合模型将真阳率提高到95.7%。图6 比较了Gu 等人的模型和Davies 等人[36]基于Goldschmidt-rule(晶体化学第一定律)预测的ABO3钙钛矿氧化物的可合成概率。

图6 ABO3钙钛矿氧化物的可合成概率对比图Fig.6 Comparison plot of synthesizability probability of ABO3 perovskite oxide
正向设计是已知结构预测性能,而逆向设计则是有了对材料性能的目标,通过算法推断出符合目标属性的材料结构,一般而言逆向设计比正向设计更有挑战性[37]。目前两种主要的逆向设计晶体材料方法包括全局优化和生成模型。全局优化指的是修改已知或者随机枚举的化合物,使用新的规则来设计新的化合物;生成模型是先从给定的数据中学习到一些隐藏的规律与分布,将训练集的数据建模,然后从中采样得到新的材料。逆向设计晶体结构的方法大都将DFT 计算与模拟退火、粒子群优化等结构搜索算法相结合,但是DFT计算代价较高。Zuo等人[38]将图卷积神经网络与对称贝叶斯优化算法相结合,先利用优化算法求得弛豫状态下的结构,然后在每个更新函数中使用3 个图卷积层进行特征提取。运用这一方法从40 万种过渡金属硼化物和碳化物中筛选出了两种新颖的硬质材料MoWC2(P63/mmc)和ReWB(Pca21),并在实验中成功合成。Cheng 等人[39]在给定23 个原子特征的情况下,生成多个随机结构,通过Voronoi分析获得相应的结构特征,然后用贝叶斯优化算法构建代理函数,再基于高斯混合模型的算法来搜索全局最小值,并将图卷积神经网络预测的形成能作为真实值迭代反馈,以此来改进贝叶斯优化模型的下一层结构。通过这一方法,成功预测了24个八重态二元化合物的晶体。
2.2 物理性质的预测
基于稳定的结构,可以利用图卷积神经网络技术预测不同结构可能具有的物理性质,包括电学性质、力学性质和热学性质等。
重要的电学性质有介电常数、介质极化率、电导率等,由于介电常数等性质的计算模拟成本较高,含有这些属性的数据集规模较小,导致训练的模型性能较差。Lee 等人[40]提出TLCGCNN方法,利用包含形成能或带隙等性质的大规模数据集进行预训练,将预训练模型卷积层中学习到的参数作为“知识”迁移到较小训练数据量的目标预测模型,以此来进行优化。结果显示,结合迁移学习时的晶体图卷积网络对介电常数预测的误差,由原始的0.181 降低至0.163。材料的电导率、热导率和超导率等重要性质与晶格振动频率有关,Nguyen等人[41]提出了更深层的基于全局注意力机制图卷积神经网络(DeeperGATGNN),针对由35,552 个晶体结构组成的Rhombohedron 数据集和由15,000 个混合类型的晶体结构组成的Mix 数据集来预测振动频率,模型在Rhombohedron 的训练集上模型训练平均绝对误差(Mean Absolute Eerror,MAE)为4.28943 Thz,测试集的MAE 为4.28879 Thz,回归模型评价指标R2为0.694,而在Mix 数据集上R2仅有0.556,这表明了在Mix数据集上的预测难度更大。DeeperGATGNN 模型的优点在于其具有可扩展性,这种可扩展性可以使网络模型的深度增加,来提取更加复杂的特征,不会像其他一些图神经网络一样,因为过度平滑而导致网络的性能下降。Laugier等人[42]将4个开源材料数据库获取的数据与Ricci等人[43]的数据集相结合,筛选清理后选取包含28 个特征的7,230 种化合物作为训练数据。晶体图卷积网络可以仅依赖组成晶体的原子信息对热电功率因数进行预测,进而得出热电性能。虽然其精度略差于全连接神经网络(Fully Connected Neural Networks,FCNN),但收敛速度更快。
材料重要的力学特征包括剪切模量、体积模量和泊松比等性质。Ahmad等人[44]使用2,041种具有剪切模量和体积模量数据的晶体结构来训练晶体图卷积网络,该模型对剪切模量和体积模量的预测结果与DFT计算值的均方根误差(Root Mean Square Error, RMSE)分别为log0.1268(GPa)和log0.1013(GPa)。为了进一步降低误差,Das等人[45]提出了一种基于自动编码器的体系结构,称之为晶体可解释特性预测器(Crystal Interpretable Property Predictor,CrysXPP)。CrysXPP 将图卷积网络用作晶体图编码器,把晶体结构以及节点和边缘的特征信息作为输入,每个节点对应的嵌入作为输出。该体系将图嵌入模块和属性预测模块相结合来预测剪切模量和体积模量,预测结果的MAE低至log0.105(GPa)和log0.08(GPa)。Levämäki等人[46]则利用迁移学习方法,先在MP数据库的有序化合物上训练了晶体图网络模型,然后将模型进行微调,利用微调后的模型在无序氮化物的数据集上预测体积模量、剪切模量和泊松比,预测精度也均在MP测试集的精度范围内。
图卷积网络不仅应用于晶体材料的电学性质和力学特性的预测,也被研究者们用于对热力学特性进行探究,如Sun 等人[47]利用AFLOW 数据库计算了5,000多种固体材料的晶格热导率和热容等热力学性质,发现大多数样品的晶格热导率介于1 到50 W/(mK)之间。运用晶体图卷积网络与一种具有提升(boosting)思想的框架相结合,将多个弱学习器联合起来形成一个强学习器。当使用整个样本进行训练,训练模型的R2(决定系数)可达0.999,MAE为0.39 W/(mK),在测试集中的MAE仅为2.747 W/(mK)。
2.3 催化反应性能的预测
除了材料本征特性的预测,图卷积神经网络还可用于预测材料的催化反应性能。催化是自然界中普遍存在的重要现象,催化作用几乎遍及化学反应的各个领域[48]。催化性能更取决于表面的结构与性质,预测不同吸附质与催化剂表面的相互作用,可以为催化剂的开发提供知道。
为了预测不同材料表面的催化能力,Chanussot 等人[49]基于MP 数据库中的体相结构,按照米勒指数面切割表面,引入吸附质,构建了开源催化材料数据集(Open catalyst 2020 dataset,OC20)。该数据集包含3 个任务:(1)给定原子位置预测吸附能和力(Structure to Energy and Forces,S2EF);(2)给定初始结构预测最终弛豫状态下原子的位置(Initial Structure to Relaxed Structure,IS2RS);(3)给定初始结构的原子位置预测弛豫结构状态下的吸附能(Initial Structure to Relaxed Energy, IS2RE)。Godwin 等人[50]提出在已有的图卷积神经网络框架上添加噪声节点作为正则化的方法,解决了网络过度平滑的问题,在IS2RE 任务中吸附能预测MAE 为0.472 eV。Shuaibi 等人[51]提出使用仅由两个原子定义的局部参照系来编码角信息,通过全局池化卷积特征后,得到的旋转卷积模型(SpinConv),不仅能够捕获丰富的角度信息,而且使体系具有旋转不变性。该方法在IS2RE 任务中吸附能预测MAE为0.437 eV。Sriram等人[52]引入图并行,将输入的图结构数据分布在多个GPU上,在OC20数据集的S2EF任务中将精度提高了15%,IS2RE任务中精度提高了21%。Gasteiger 等人[53]通过在几何消息传递神经网络(GemNet)[54]的基础上额外对原子间距离、相邻边角度以及二面角角度编码,运用该模型进一步将预测性能提高了16%,同时将训练时间减少为原来的1/10。近期,Korovin等人[55]先利用Voronoi图对结构进行分区确定节点领域,然后在编码过程中分别在原子嵌入中加入键角、键类型,元素属性等信息,此时训练出来的模型在OC20数据集中的误差仅为651meV。Sriram 等人[56]则将DimeNet++和GemNet 模型的参数量扩大了一个数量级,将其输入运用图并行在多个gpu 进行训练,在OC20数据集的S2EF 任务上预测力的精度提高了15%,在IS2RS任务上预测性能提高了21%。
Kim 等人[57]开发了一种表面图卷积神经网络(Slab Graph Convolutional Neural Network,SGCNN),将DFT 计算的3,040 个表面数据作为数据集,预测了与氮还原反应(Nitrogen Reduction Reaction,NRR)性能相关的5种吸附质的结合能,其MAE值为0.23 eV。Back等人[58]修改网络结构后,使得网络能够适应初始结构中处于任意位置的吸附质原子,通过利用初始结构及其与最终结构之间的距离变化,预测纯金属、化合物和合金不同表面CO 和H 结合能,预测的MAE为0.15 eV。Tran等人[59]基于MP数据库构建了大量表面模型,探索用于二氧化碳还原的催化剂。这一工作构建了包含近40,000个结构的数据集,利用Back等人[58]的方法进行训练的MAE为0.19 eV。Gu等人[60]在Tran等人的数据集中标记了未经过弛豫计算的原子结合位点,以此数据集为基础,提出了一种减少模型偏差的集成学习方法,预测了结合能的概率分布。运用此模型测试CO 和H 的结合能的MAE 分别为0.116 eV 和0.085 eV。Li 等人[61]则利用Kevin Tran 的数据集,将图神经网络算法[62-63](DimeNet++)与Gu的方法[60]结合,对低成本的二元/三元铂合金析氢催化剂进行预测以此来寻找高性能但高成本的铂催化剂的替代品,结果表明在二元合金中Cu3Pt(100)的吸附性能与Pt(111)几乎相同,H、CO 和O 吸附能的最大偏差为0.04 eV;在三元合金中FeCuPt2可以成为Pt 的低成本替代品,该催化剂的(100)面以及(001)面在H、CO 和O 的吸附方面表现出与Pt(111)相似的性能(最大偏差为0.16 eV)。Wu等人[64]从2D MatPedia数据库中筛选了分解能小于0.1 eV且无带隙的二维材料,进行结构优化后,选取其中稳定的455 种材料,考察所有的活性位点。经过图卷积层和池化层对晶体结构进行变换后,利用全连接层学习参数预测吸附能,判断材料能否能成为好的催化剂,并最终从3,599种复合材料中筛选出38种稳定性较好的潜在析氢催化剂材料。
上述大部分图卷积网络都是单纯的基于数据驱动的回归模型,Wang 等人[65]则提出了一种将理论注入神经网络的方法(Theory-Infused Neural Network,TinNet),将图卷积网络与物理化学理论相结合,架构如图7所示。这一方法分为回归模块和理论模块,将吸附-基底系统的特征表示作为回归模块输入,回归模块的输出单元作为理论模块的未知参数,运用理论模块来预测金属位点的吸附特性。运用该模型预测合金表面的OH 的化学吸附能,MAE 为0.118±0.022eV,小于全连接神经网络模型的预测误差(0.152±0.015eV)。该体系框架有望构建能准确预测活化能、吸附质相互作用性质的模型,帮助筛选反应中间体更稳定的催化剂。

图7 TinNet网络架构Fig.7 TinNet architecture
3 总结和展望
本文简要介绍了图卷积神经网络的背景原理,目前常用的图卷积网络方法分为基于频谱的方法和基于空间的方法:基于频谱的方法通过将节点域转化为频域、再映射回节点域的方法来定义图卷积;基于空间的方法则通过寻找中心节点并聚合其邻居节点的特征信息来定义图卷积。后者近年来在晶体材料的研究中得到了大量应用。本文进而对图卷积神经网络在晶体材料的结构设计和性能预测方面的典型应用进行了介绍,目前应用在晶体材料领域效果较好的网络大都将晶体结构中的原子和键分别用节点和边表示。将元素类型和键距、键角等信息进一步编码到节点和边的属性中。网络基于图结构通过相邻节点及其边之间的卷积操作来更新节点级、边级和全局的嵌入,然后对此可学习的过程进行训练,来预测所研究材料的特定属性。这些应用也显示了图卷积神经网络相较于传统的密度泛函理论计算具有代价低、耗时短的特点。数据是神经网络建模的基础,图卷积神经网络预在材料领域中得到更好地应用亟需完善和扩大探究材料领域中典型问题所需的数据规模,在高质量数据的保证下建立新颖、合理且高效的网络架构,选择机器更易学习的特征表示将其输入模型来预测材料属性,基于此些必备条件,可望更快实现数据驱动的材料设计。但是该方法目前也存在以下挑战:
(1)如何提高模型的泛化能力以及如何评判各个SOTA 模型的泛化能力大小。目前多数图卷积神经网络在训练数据集和测试数据集上的效果较好,但是在对外部数据进行预测时,模型预测结果的准确性难以保证。大部分机器学习方法为了提高模型的泛化能力一般从两方面着手。第一个研究方向是从数据出发,通过扩充训练集数量、数据增强、标注数据更易学习的特征等方法。第二个研究方向是从模型出发,在模型构造时,通过增加模型的深度与宽度;在模型训练时,通过融合dropout[66]、早停法、交叉验证[67-68]、迁移学习或者选择更优的损失函数、优化器[69-71]、调节批处理尺寸等方法;在模型训练完后进行后处理,例如各种集成学习[72-73]的方法。相比卷积网络适用的图像等数据,图结构的材料数据较难对数据进行增强与标注,因此若欲从数据出发提高图卷积网络在材料领域的泛化能力,则要考虑扩大数据集的规模或者通过GAN等方法间接按照需求对数据进行特定的处理。若欲从模型的角度出发,使用新颖的交叉验证方法[74],寻找网络适宜的评价函数、损失函数[75-77],添加不同的正则化方法等可能更易提高图卷积网络在材料领域的泛化能力。如果想要对目前各个材料领域SOTA 模型的泛化能力进行评估,需要满足一些标准,如:①使用相同并且能代表材料化学中典型问题的数据集对模型进行评估;②统一模型对数据集中编码的信息;③在各个模型训练优化时选择相同的超参数。在控制变量的条件下才可以真正判断各个领域的SOTA模型真正的泛化能力,由此才可进一步推断哪些模型能更好地为材料专家提供有效帮助。因此迫切需要进行如Fung等人[78]所做的工作:建立一个评判的基准框架。
(2)如何加深网络的结构,以提取深层特征。大部分传统的卷积神经网络是通过堆叠卷积层来得到细粒度模型。但是对于图卷积神经网络来说,“卷积”操作是针对各个节点和边信息的聚合和更新的过程,因此如果增加图卷积层的数量会引入噪声易造成节点平滑,导致网络性能下降,因此如何在提取到深层特征的前提下,减小噪声对模型的影响是GCN的瓶颈。
(3)如何提升网络的可拓展性,使得网络更加“通用”。目前GCN在材料领域研究所采用的数据,一部分来自在线数据库,一部分来自已发表的论文,还有部分来自高通量实验数据。不同的数据来源导致数据规模、质量等多方面的差异。某个特定材料领域数据集所含有的目标性能规模较小,导致“通用”模型无法很好地学习到深层特征。
利益冲突说明
所有作者声明不存在利益冲突关系。
