聚合物分子动力学软件PyGAMD及在国产异构计算平台的适配
2024-02-27于卓辰蒲鑫韩秉豫朱有亮吕中元
于卓辰,蒲鑫,韩秉豫,朱有亮*,吕中元
1.吉林大学,化学学院,超分子结构与材料国家重点实验室,吉林 长春 130012 2.河南师范大学,软件学院,河南 新乡 453007
引 言
分子动力学模拟是研究微观原子和分子集体行为的重要方法,通过积分经典运动方程在相空间抽样,进一步计算体系热力学量的系综平均及其他性质。该方法是对理论和实验的有力补充,也用于预测目前无法用实验方法表征的现象,直观地展示了实验现象发生的机理与规律[1-4]。分子动力学模拟在医药研发、纳米材料、金属材料和软物质材料等研究领域发挥着关键作用。常见的分子动力学模拟软件有:瑞典皇家理工学院主导开发的GROMACS[5];美国桑迪亚国家实验室开发的LAMMPS[6];伊利诺伊大学香槟分校开发的NAMD[7];加利福尼亚大学开发的AMBER[8-9]以及哈佛大学开发的CHARMM[10]。
聚合物材料应用非常广泛,它和金属、无机材料并称为人类文明的三大支柱材料。聚合物是典型的软物质,行为复杂多样,一直是计算机模拟仿真研究的重点。由于聚合物分子链较长,其结构与动力学的多尺度特征给分子模拟研究带来了严峻的挑战。分子模拟的全原子模型考虑了全部原子级别的细节,其巨大的计算量限制了模拟尺度(通常模拟尺寸在几纳米到几十纳米量级,模拟时间小于100纳秒),在研究聚合物结构和动力学性质方面非常受限。通过忽略掉一些无关紧要的分子细节,一个粒子代表一个原子团的粗粒化模型可以大大提高模拟效率。
我们基于聚合物的粗粒化模型,发展了多个行之有效的模拟模型与方法,如动态键模型、聚合反应模型、定制数值势模型、各向异性粒子模型、粒子-密度场方法、快速静电计算方法以及静电耗散粒子动力学方法等。通过结合上述粗粒化模型/方法和高性能计算,我们开发了聚合物分子动力学模拟软件GALAMOST[11-12],使得聚合物体系的模拟研究能够达到微秒和微米的时间和空间尺度。GALAMOST 作为独立自主开发的国产分子动力学模拟软件,同时被NVIDIA 官网应用目录和SklogWiki 材料模拟程序目录收录。它利用GPU 实现了高性能计算,是计算效率最高的分子动力学模拟软件之一,主要适用于聚合物、生物大分子、胶体粒子、液晶等软物质体系的结构和动力学性质研究。目前该软件在多个领域得到了广泛应用,如复旦大学的聂志鸿等采用聚合反应模型和静电计算方法,描述纳米粒子接枝分子链间的可逆反应和静电排斥作用,解释实验上利用纳米粒子构筑“胶体分子”的内在机理[13];南洋理工大学的M.P.Ciamarra 等利用具有不同势能形式的胶体粒子模型,阐释了其二维体系从连续到不连续相结构转变的机理[14];福州大学的宋继彬等利用各向异性粒子模型解释实验上高分子接枝纳米粒子形成双层囊泡的自组装机理[15];上海交通大学的陆学民等利用耗散粒子动力学模拟阐明螺旋线调控的嵌段共聚物薄膜结构中螺旋效应的影响因素[16]。
我们开发聚合物分子动力学模拟软件主要采用Fortran、C++和CUDA C 等编程语言,通过合理设计的程序架构实现了软件的模块化和拓展性。为了提高计算效率,我们基于CUDA C发展了分子动力学模拟的GPU 加速算法,软件中全部运算由GPU完成,CPU和GPU之间不需要频繁的数据通信。GPU含有数千至上万计算核心,与CPU相比,同价位GPU的浮点计算性能是CPU 的10~100 倍,在分子动力学模拟方面具有较高的性价比。但是GPU要求计算任务具有很高的并行性,即计算任务需要能够很好地分解为众多独立的子并行计算任务。GPU计算效率依赖于众线程算法,对高度依赖数据交换的分子动力学模拟来说是个很大的挑战。为此,我们发展了一个线程负责一个粒子的GPU并行策略,并在此基础上发展了一系列动力学模拟方法的GPU 算法。此外,我们还利用近邻粒子搜索算法和希尔伯特数据整理算法等进一步提升了软件的计算效率。
目前,编译器的发展已经到了分子动力学模拟程序功能与效率协调的极限。如何通过软件形式创新解决功能与效率的矛盾问题,是发展高性能分子动力学模拟软件的未来出路。解释器(Interpreter)是一种用于直接执行源代码的计算机程序。与编译器不同,解释器不会将源代码完全转换为机器码,而是逐行读取源代码,并逐行解释执行。虽然解释器通常要比编译器的执行速度慢,但是解释器更加灵活和便捷,研究人员可以立即看到代码更改的效果,并且能够实现一些编译器无法实现的功能,特别适合程序的快速开发和多功能的高度集成。Python是一种解释型语言,其源代码通过Python 解释器执行。Python的语法简洁清晰,具有强大的标准库和第三方库,配合如NumPy、Numba、SciPy和Pandas等可以实现高效的科学计算和数据分析,并且可以跨平台兼容,在多种操作系统上运行,增强程序的可移植性。此外,Python 支持Tensorflow 与PyTorch 等主流深度学习框架,是目前人工智能与机器学习领域主要使用的语言。因此,基于Python 开发一款分子动力学模拟解释器,使用户方便地自定义应用模块,对降低分子模拟领域中定制化功能的开发难度有着重要意义。
1 分子动力学模拟解释器PyGAMD
目前分子模拟领域仍充满挑战,如特殊体系的势函数开发[17]、自定义积分方法[18]、机器学习力场[19]等。传统的分子动力学模拟软件主要采用Fortran、C 或C++这些编程语言构建,这导致了其扩展能力受限,使得定制化分子模拟成为一项挑战。复杂的软件架构使得普通用户几乎无法添加新的功能。然而,解释性编程语言的兴起为软件设计带来了全新的思路。因此,借助解释性语言的灵活性和易扩展性,重新构建软件框架,以便于支持二次开发和增添新功能,已经成为一个迫切的需求。此前,由于Python 计算效率低下而一般不采用Python 进行开发,对于用户自定义分子动力学模块(如势能函数形式,积分方法等)时,用户需要自行改写分子模拟软件的源码并重新编译,还需熟练掌握CUDA的开发环境以提高软件的运行效率,用户自行开发的难度极高。
因此,我们基于Python开发了分子动力学解释器—PyGAMD(Python GPU-Accelerated Molecular Dynamics Software; 网址http://pygamd.com/)。PyGAMD 是一款基于Python 语言和Numpy、Numba 等扩展库编写的高扩展性、高自定义性的GPU加速分子动力学解释器。用户通过简单的示例即可迅速掌握自定义分子动力学模拟功能的方法,并实现高度定制化的模拟功能需求。基于Python语言接口的灵活性与扩展性,用户可以将多种外部库与PyGAMD 联合使用,具有便捷、高效和语法清晰等特点。在PyGAMD 中,用户既可以基于给定的软件框架实现自定义势能和积分函数等功能,还可以通过Python的Numba库来进行GPU加速的分子动力学模拟新功能开发。Numba 是一个开源的即时(Just-In-Time,JIT)编译器,它使用LLVM编译器基础结构将Python 函数转换为优化的机器码。Numba允许用户使用Python语言来编写高性能的科学计算函数,通常只需要在函数上添加装饰器(decorator),不需要较高的学习成本。同时,Numba 支持GPU 代码的开发,与直接使用CUDA C相比,使用numba.cuda.jit编写GPU代码通常更简单,更易于学习,并且无需离开Python的开发环境。
1.1 PyGAMD程序架构
PyGAMD 解释器程序架构如图1 所示,通过设置二级结构,将进行一次完备分子动力学模拟所需的各个部分组合起来,传递给主对象Application。运行分子动力学模拟需要调用Application 对象中的run 函数,用户需要将分子动力学模拟所必要的部分传给Application 对象,包括文件的读取(一级结构Snapshot,二级结构各文件类型读取方法)、力的计算(一级结构Force,二级结构成键/非键势能等)、积分器(一级结构Integration,二级结构为各类积分器)与输出构型文件(一级结构Dump,二级结构为各类文件格式)。其中,Force 和Integration 对象中大量使用GPU 函数,PyGAMD 允许用户在计算脚本中直接通过numba.cuda.jit自定义所需的函数。

图1 PyGAMD的程序框架Fig.1 Program framework of PyGAMD

图2 PyGAMD软件的组成Fig.2 Composition of the PyGAMD
此外,我们还通过把PyGAMD 的接口进行拓展,囊括了多个自研的外部程序工具,包括聚合物建模工具库molgen,异构加速库cu_gala/hip_gala 和数据处理工具dataTackle 等,使得PyGAMD 成为一个集聚合物建模、动力学模拟和数据分析为一体的聚合物分子动力学软件平台。
1.2 平台支持情况
目前,PyGAMD可以在Linux和Windows平台安装和使用,平台要求:Intel/Amd CPU,NVIDIA GPU 或国产异构加速卡,python 3.11 与CUDA Toolkit(>=12.2)。PyGAMD软件支持离线和在线安装,离线安装可以通过网站上的地址找到GitHub 托管的开源软件项目,下载软件包并通过一条命令安装(如下第一条命令),在线安装需要连接外部网络,并通过一条命令进行自动下载和安装(如下第二条命令):
对于国产异构加速计算平台,由于目前还未支持Numba 库,PyGAMD 的自定义功能使用受限,但仍可通过调用hip_gala库来实现高效的分子动力学模拟,并利用数值势能方法实现自定义势能函数功能,如下图代码所示:

1.3 PyGAMD的使用
1.3.1 用户自定义力场
用户可在计算脚本中通过Numba直接定义所需要的力场势能函数和参数,只需要简单地在核函数前加入numba.cuda.jit decorator即可方便地定义GPU函数。譬如自定义Harmonic bond作用势:

除了键伸缩作用势外,自定义函数还支持非键作用势、键角弯曲作用势和二面角扭转作用势等。势能函数中的参数个数和含义由用户指定,共同构成了分子模拟的力场。
1.3.2 异构加速库
PyGAMD 集成了异构加速库cu_gala 与hip_gala。对于NVIDIA 的GPU 可以在脚本中加载cu_gala 库,国产异构加速计算平台需要在脚本中切换为hip_gala,具体的加载命令如下:

我们利用PyGAMD异构加速库完成了一些应用测试,很好地复现了我们之前的分子动力学模拟结果,比如利用布朗动力学方法模拟多臂星形嵌段共聚物(Ax1)y-C-(Bx2)y 的自组装,复现了多臂星形嵌段共聚物在溶液中的自组装结构与分子链长和溶剂性质参数α的相图[20];利用软的各向异性补丁粒子模型,复现了二维开放晶格形成的动力学[21];利用粗粒化模型复现了能量耗散的超分子动态自组装和解组装过程[22];利用粗粒化分子动力学模拟,复现了常温常压自发相分离纺丝过程,对溶质/溶剂在时间和空间上的传递过程进行了定量化描述[23],确认了异构加速库计算的精准性。
1.4 异构加速库计算效率
为了检测PyGAMD 在各计算平台上的计算效率,我们分别在WSL2-3060 (GPU)、GTX 1080(GPU)以及国产异构计算加速卡上,使用异构加速库对五种常见分子模拟体系进行计算,统计其计算效率(图3)。这五种体系分别为:双嵌段共聚物相分离——DPD-A5B5,双嵌段共聚物相分离——DPD-A3B7,MARTINI 力场下对DPPC双分子膜的模拟——MARTINI-DPPC,利用粒子-密度场方法研究POPG 双分子膜——MDSCF-POPG,各向异性粒子的自组装——Gay Berne Model。
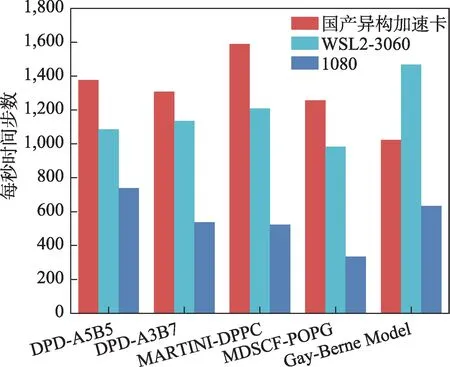
图3 PyGAMD异构加速库在国产异构加速卡、WSL2-3060(GPU)、GTX 1080(GPU)运行算例的计算效率一览Fig.3 Overview of computational efficiency of PyGAMD heterogeneous acceleration library running examples on domestic heterogeneous acceleration unit,WSL2-3060(GPU),GTX 1080(GPU).
在五种测试体系中,异构加速库计算效率均表现优异。其中,国产异构加速卡在大多数算例中的表现优于在WSL2-3060(GPU)中的表现。只有各向异性模型——Gay Berne Model在国产卡上的计算效率低于WSL2-3060(GPU),这可能是国产异构加速卡独特的硬件架构所致,后续将针对国产异构加速平台对代码进一步调优,以提升各方面的计算效率。
1.5 PyGAMD在国产异构加速卡上的适配

1.6 PyGAMD未来发展的展望
经典分子动力学(classical molecular dynamics)模拟依赖于精确的物理模型来计算分子之间的相互作用。对于复杂体系(如生物大分子与高分子聚集态结构等),目前基于经验或DFT 拟合的力场参数的精确性仍有较大的提升空间。因此,人们开始寻求新的方法来加速分子模拟和解决这些问题。机器学习提供了一种从已有数据中预测新数据的方法。在分子动力学模拟中,机器学习方法可以用于快速估计分子之间的相互作用势能曲线,或者用于学习分子之间的复杂相互作用。目前主流的机器学习(scikitlearn)与深度学习框架(Tensorflow,PyTorch)均基于Python,PyGAMD作为基于Python的分子动力学模拟解释器,在对接相应的机器学习势[24]时具有优势。我们后续将开发相应模块与接口(图4)并注重基于机器学习的粗粒化模拟方法的开发。
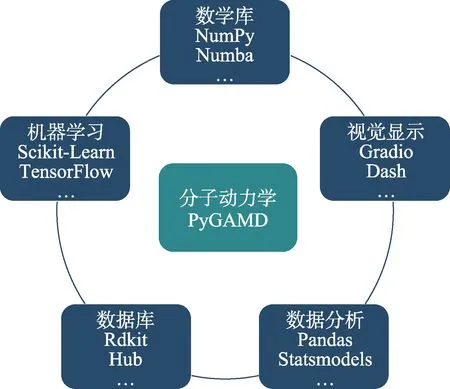
图4 PyGAMD具有较强的扩展性Fig.4 PyGAMD is highly scalable
此外,虽然PyGAMD 在粗粒化模拟中具有效率高与扩展性强等特点,但是由于粗粒化模型缺少原子细节,无法进行一些材料性质计算(如光电、介电、吸附等)。为此,我们拟通过开发细粒化(Fine-Grained)方法将粗粒化模型映射为全原子模型,同时发展增强采样算法生成低能态的局部构型、空间自规避算法体现原子的体积排除作用并防止键穿透、能量最小化算法实现结构优化等,进而在模拟过程中实时地将粗粒化模型转化为全原子模型,构成全原子模型演化轨迹,从而进行多种物理性质的精确计算。
在人工智能时代,深度学习框架有助于加速复杂的数值计算问题,甚至绕开数值计算本身,直接得到所需结果。传统分子动力学模拟往往将力的来源追溯为势能的负梯度,这使得科学家们不断地去精进经典力场的精度与泛化能力。但是,经典力场往往只能通过有限项的线性相加来尽量贴合实验值或电子结构计算模拟值,如将力场构建为成键作用势与非键作用势(范德华作用与经典)的加和。但显然,再精确的经典力场,由于其参数非灵活,实则永远无法得到与真实体系相一致的参数空间。所以,科学家提出一系列计算方法,来实现利用电子结构计算所产生的势能面求得原子受力的方法,进一步应用于分子动力学。但是,即便是效率较高的CPMD 算法,也将耗费大量的核数及长达数周或数月的时间来产生Picosecond 级的结果。为此,科学家提出利用深度学习,将电子势能面及位置作为输入,来预测新的原子坐标下的电子势能面,这样无需每一步迭代都计算一次DFT 级别的电子势能,极大地节约了计算时间。我们计划将PyGAMD 与VASP、CP2k 等第一性原理软件对接,利用DFT 产生的电子势能面进行力的计算,之后尝试引入深度学习框架,实现机器学习加速的第一性原理分子动力学模拟。
此外,在数据驱动时代,实验人员从希望理论计算模拟验证材料性能与机理,变为了希望从模拟出发预测优选性能的材料。为此,构建高通量计算工作流以优选材料,是PyGAMD 后期的关键目标之一。一个完整的高通量筛选模块包括自动模型搭建、自动性能计算、自动后处理以及自动训练及预测等功能,在自动搭建以及自动预测功能中,PyGAMD 有望引入Pytorch等工具协助生成全原子模型,并为其分配最合适的力场参数与电荷。以及能够通过用户输入的特征来进行自动训练,并从数据库中筛选所需性能的材料。用户可根据神经网络的预测结果来进行实验合成,完成闭环高通量筛选的工作流程。
2 总 结
PyGAMD 是基于Python 语言,使用Numba等扩展库,构建的一款高扩展性、高自定义性、跨平台的GPU加速分子动力学模拟解释器。用户可以轻松掌握自定义分子动力学模拟模块的方法,并满足高度定制化的功能需求。用户可以将PyGAMD与其他Python库结合以实现复杂的计算模拟流程。传统上,分子模拟领域中定制化功能的开发难度较高,通过PyGAMD,用户无需改写复杂的C++或Fortran 源码或掌握CUDA C 开发环境,大大降低了定制化功能的开发难度。此外,PyGAMD 作为基于Python 的解释器,在对接主流的机器学习和深度学习框架(如scikit-learn、Tensorflow、PyTorch 等)方面具有优势,这有助于推动基于机器学习的通用粗粒化方法的开发。PyGAMD解释器的出现不仅为分子动力学模拟领域的研究人员提供了强大、灵活的工具,还推动了分子模拟技术与高性能计算技术和解释器技术相结合,为未来的科学研究和创新打开了新的窗口。
利益冲突声明
所有作者声明不存在利益冲突关系。
作者贡献声明
于卓辰,蒲鑫与韩秉豫对本文章的贡献相同。于卓辰:撰写和汇总;蒲鑫:撰写和文献综述;韩秉豫:软件调试和测试;朱有亮:文章修改;吕中元:文章修改。
