液液萃取/气相色谱-串联质谱法测定人尿中16种羟基多环芳烃
2024-02-27钱柬坤陆一夫
鲍 珊,钱柬坤,2,付 慧,邱 天,陆一夫,3*
(1.中国疾病预防控制中心环境与人群健康重点实验室,中国疾病预防控制中心环境与健康相关产品安全所,北京 100021;2.中国医科大学 公共卫生学院,辽宁 沈阳 110122;3.南京医科大学 公共卫生学院全球健康中心,江苏 南京 211166)
多环芳烃(PAHs)是一类广泛存在的环境污染物,主要来自化石能源燃烧、汽车尾气排放、烟草燃烧、食物加工等过程中有机物的热解或不完全燃烧,通过消化道摄入、呼吸道吸入和皮肤接触等途径进入人体[1-4]。PAHs 暴露引起的健康风险已受到国际上广泛关注,国际癌症研究中心(IARC)的研究证实PAHs是一类具有致癌、致畸、致突变毒性的化学物质[5-7]。根据其在环境中的分布、人群暴露和健康风险,美国国家环境保护局(USEPA)将萘、芴、菲、芘、荧蒽、苯并[a]芘等16种PAHs列入优先控制污染物名录[8],我国原环境保护部会同工业和信息化部、卫生健康委将萘、蒽、苯并[a]蒽、苯并[a]菲、苯并[a]芘、苯并[b]荧蒽等8种PAHs列入《优先控制化学品名录》。
PAHs 进入人体内经细胞色素P450 催化反应生成羟基化代谢产物(OH-PAHs),最后通过尿液排出体外[6,9]。目前研究表明人尿中OH-PAHs 含量能够全面地反映PAHs 的暴露水平,美国和加拿大已对普通人群尿中OH-PAHs 负荷水平和变化趋势进行长期监测[10],我国2016 年起组织实施的国家人体生物监测项目也将尿中OH-PAHs纳入监测指标[9]。
目前测定尿中OH-PAHs常用的前处理方法主要有液液萃取(LLE)和固相萃取(SPE)[4,11],LLE因其成本较低、操作简便等优点更适用于大批量样品的检测。液相色谱-质谱联用法(LC-MS)和气相色谱-质谱联用法(GC-MS)是常用于尿中OH-PAHs的仪器分析方法[12-15]。LC-MS具有无需衍生化且操作步骤相对简单等特点,但此类方法无法实现2-羟基芴和9-羟基芴、2-羟基菲和3-羟基菲等同分异构体的分离,同时存在较强的基质效应和灵敏度相对较差等问题[16-17]。GC-MS相比LC-MS 具有更好的分离能力,且气相色谱-串联质谱(GC-MS/MS)的多反应监测模式(MRM)可进一步提高羟基荧蒽、羟基苯并[a]芘和羟基苯并[a]蒽等痕量目标物的灵敏度[1,11,18-20]。
本研究通过对实验条件的优化选择,建立了一种可同时测定人尿中16 种OH-PAHs 的液液萃取/气相色谱-串联质谱法(LLE/GC-MS/MS)。该方法已应用于我国国家人体生物监测项目中,通过监测人群尿中OH-PAHs负荷水平和变化趋势,可全面评估PAHs暴露对人群健康的影响。
1 实验部分
1.1 仪器、试剂与材料
Trace 1310-TSQ 9000 气相色谱-串联质谱仪(美国Thermo Fisher 公司),TG-5SilMS 色谱柱(美国Thermo Fisher 公司);FV64 氮吹仪(广州得泰仪器科技有限公司),DT5-4B 离心机(河北医众医疗器械有限公司)。
16 种OH-PAHs 标准溶液(天津阿尔塔科技有限公司,质量浓度均为100 mg/L),10 种13C 标记的同位素定量内标溶液和回收内标(美国Cambridge Isotope Laboratories 公司,质量浓度均为50 mg/L),4 种氘代标记的同位素定量内标标准物质(纯度>98%,加拿大Toronto Research Chemicals 公司),其详细信息见表1。正己烷(农残级,美国Fisher 公司),正戊烷(色谱纯,德国默克公司),甲苯(色谱纯,美国Tedia 公司),正十二烷(色谱纯,比利时Acros 公司),β-葡萄糖苷酸酶/硫酸芳酯酶(β-葡萄糖苷酸酶活度单位≥300 000 U/g,硫酸芳酯酶活度单位≥10 000 U/g,德国默克公司),N-甲基-N-(三甲基硅烷)三氟乙酰胺(MSTFA)、双(三甲基硅烷基)三氟乙酰胺(BSTFA)(色谱纯,美国Sigma-Aldrich 公司),其他试剂均为分析纯。实验用水均为纯水。
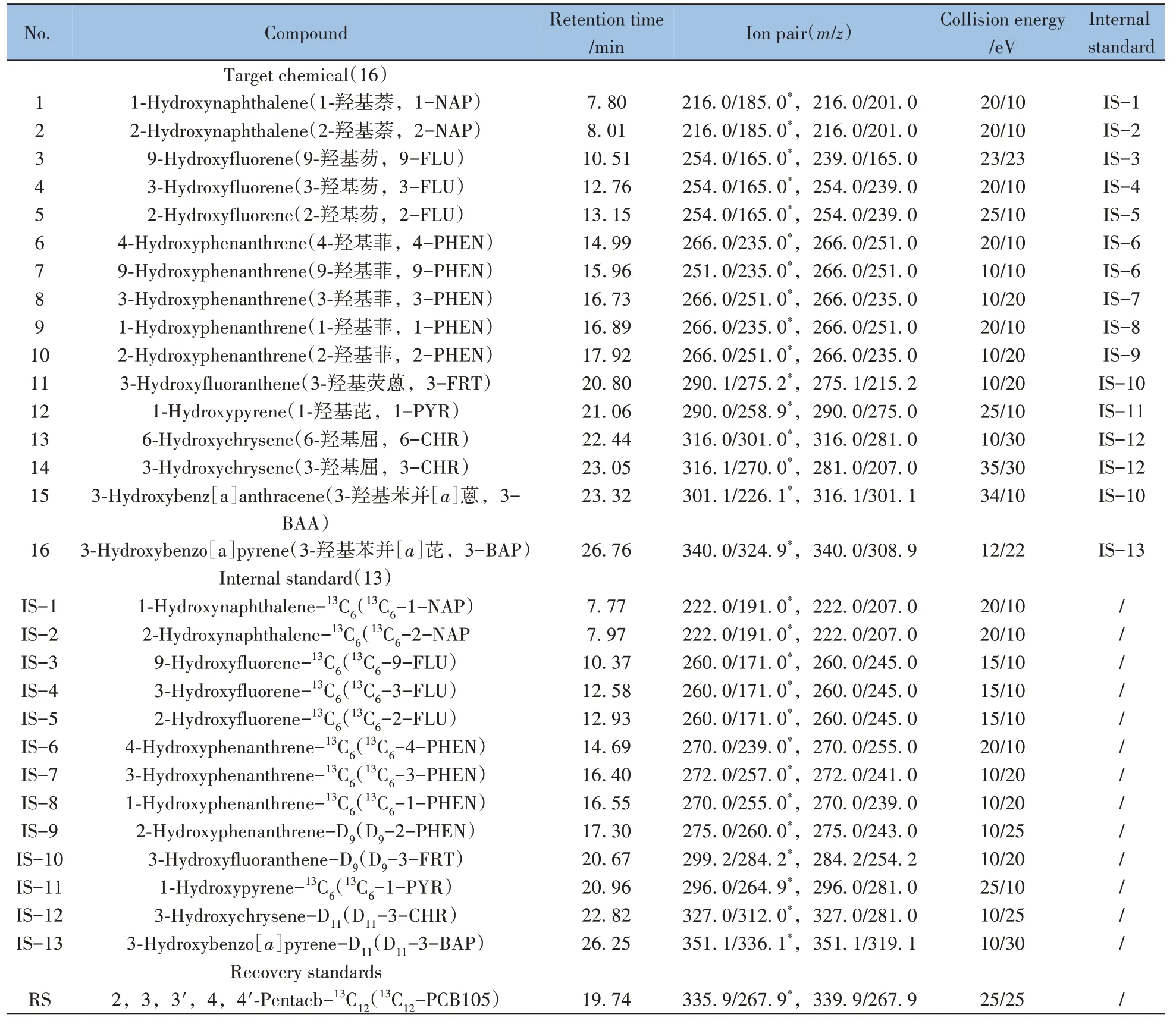
表1 16种羟基多环芳烃及其同位素内标的仪器分析参数Table 1 Instrument analysis parameters of 16 OH-PAHs and their internal standards
1.2 溶液配制
1.2.1 抗坏血酸溶液称取适量抗坏血酸用水溶解稀释为0.25 mg/L的抗坏血酸溶液。
1.2.2 硝酸银溶液称取适量硝酸银用水溶解稀释为1 mol/L的硝酸银溶液。
1.2.3 酶溶液称取适量无水乙酸钠用水溶解稀释为1 mol/L 的乙酸钠溶液(乙酸调至pH 5.5),再取适量β-葡萄糖苷酸酶/硫酸芳酯酶溶于乙酸钠溶液中得到β-葡萄糖苷酸酶浓度为3 000 U/mL,硫酸芳酯酶浓度为100 U/mL的酶溶液。
1.2.4 标准使用溶液用甲苯分别稀释配制质量浓度为1 mg/L 的混合标准使用溶液(1-羟基萘和2-羟基萘的质量浓度为5 mg/L),质量浓度为25 μg/L 的混合同位素定量内标使用溶液,以及质量浓度为100 μg/L的回收内标使用溶液。
1.3 尿样采集
本研究依托国家人体生物监测项目共采集529 名调查对象的尿样,该项目已获中国疾病预防控制中心环境与健康相关产品安全所伦理委员会批准(批准号:201904)。
1.4 样品前处理
1.4.1 酶解尿样解冻后取1 mL 于12 mL 玻璃管中,依次加入80 μL 定量内标使用溶液、10 μL 抗坏血酸溶液和1 mL酶溶液后振荡混匀,于37 ℃下避光酶解至少12 h。
1.4.2 液液萃取酶解液中加入2 mL水后,加入5 mL甲苯-正戊烷(1∶4)振摇10 min后,3 500 r/min离心10 min,取上层有机相转移至另一玻璃管中,酶解液中再加入5 mL 甲苯-正戊烷(1∶4)重复提取一次,合并两次提取的上层有机相后加入1 mL 硝酸银溶液,振摇5 min 后,3 500 r/min 离心5 min,取上层有机相于60 ℃氮吹至近干。
1.4.3 衍生化向氮吹试管中加入20 μL 甲苯复溶后转移至已加入10 μL 回收内标使用溶液和10 μL MSTFA的棕色进样小瓶中,震荡混匀后,60 ℃避光放置50 min后待测。
1.5 仪器分析条件
气相色谱条件:进样口温度:300 ℃;进样方式:不分流进样;载气(氦气)流量:恒流模式,0.9 mL/min;进样量:1 μL;升温程序:起始温度95 ℃,保持1 min,以15 ℃/min 升温至195 ℃,再以2 ℃/min升温至206 ℃,保持5 min,再以40 ℃/min升温至300 ℃,保持8 min。
质谱条件:EI 离子源温度为300 ℃;传输线温度为300 ℃;多反应模式(MRM)扫描,碰撞气为氩气,溶剂延迟时间5 min。
2 结果与讨论
2.1 衍生化反应条件的选择
由于目标物中含有极性较强的羟基官能团,易与气相色谱柱填料相互作用形成氢键,影响色谱柱的解吸能力,因此在仪器分析前需通过甲硅烷基化反应取代羟基进行衍生化以提高分析灵敏度[21]。MSTFA 和BSTFA 是最常用的衍生化试剂,已有研究表明基于MSTFA 进行甲基硅烷化生成的衍生物可获得更高的丰度,因此本研究使用MSTFA 作为衍生化试剂[22]。衍生化反应的温度和时间是影响结果的主要因素,既往研究根据反应速率和化学平衡证实60 ℃是最佳温度[19,23]。本研究在此温度条件下比较了基质样品目标物质量浓度为2 μg/L(1-羟基萘和2-羟基萘为10 μg/L)时不同反应时间(20、30、40、50、60、70 min)对实验结果的影响。结果表明,当衍生化反应时间为50 min 时目标物响应达到峰值。因此本研究选择衍生化反应时间为50 min。
2.2 仪器条件的优化
本研究使用常用的95%二甲基聚硅氧烷(5%二苯基)固定相填料的TG-5SilMS(30 m×0.25 mm×0.25 μm)弱极性色谱柱,通过优化进样口温度、柱流速、升温程序等色谱条件,确保各目标物的分离度良好。图1为16种目标物的色谱图。
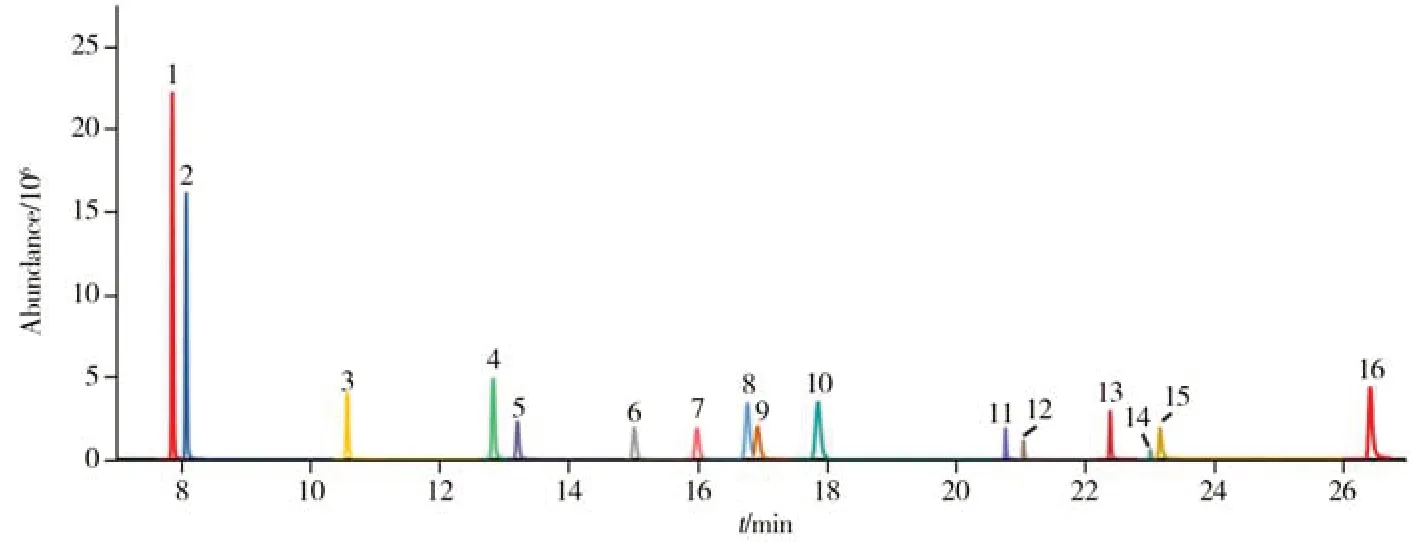
图1 16种OH-PAHs混合标准溶液的色谱图Fig.1 Chromatogram of 16 OH-PAHs mixed standard solution the numbers denoted are the same as those in Table 1;mass concentrations of 14 OH-PAHs were 20 μg/L,those of 1-NAP and 2-NAP were 100 μg/L
采用多反应监测模式(MRM)对标准溶液进行全扫描,以目标物三甲基甲硅烷基衍生物的分子离子为母离子确定保留时间,根据裂解规律选择M-30(-2[·CH3])、M-31(-[·CH3+ CH4])和M-89(-OSiC3H9)等质荷比大且响应值高的特征基团作为其产物离子[1,19,21],得到3~5个备选离子对组合,再进一步优化其碰撞能量。由于尿样基质复杂,为避免实际样品检测过程中离子对的基质干扰,按照“1.4”和“1.5”的方法测定基质加标样品,确定最优的定量、定性离子对。最后优化并确定传输线和离子源温度等质谱参数。仪器条件参数见表1。
2.3 酶加入量的选择
OH-PAHs在尿中主要与葡萄糖苷酸和硫酸盐以偶联形式存在,因此测定时首先需要通过酶水解法将目标物从偶联结合态转化为游离态后再进行提取[24]。β-葡萄糖苷酸酶/硫酸芳酯酶加入量是影响酶水解效率的关键因素,一般酶解效果随着酶加入量的增加而提升。本研究比较了质量浓度为2 μg/L(1-羟基萘和2-羟基萘为10 μg/L)基质样品选择3种不同酶加入量(1 500(50)U、3 000(100)U和4 500(150)U)时对目标物响应强度的影响。结果表明,当β-葡萄糖苷酸酶/硫酸芳酯酶加入量达到3 000(100)U 时,目标物的响应强度达到峰值。因此本研究选择1 mL 尿样中加入3 000(100)Uβ-葡萄糖苷酸酶/硫酸芳酯酶。
2.4 提取溶剂的选择
OH-PAHs 的pKa值为8.0~9.2,这类弱极性有机物通常采用非极性或极性较小的溶剂进行提取[12]。本研究比较了体积均为10 mL 的正己烷、正戊烷和甲苯-正戊烷(1∶4)3 种不同提取溶剂的提取效率(ER),按照“1.4”步骤处理空白基质加标样品,通过公式ER(%)=A1/A2×100%对结果进行评价,其中A1为样品前处理前基质加标测得的目标物定量离子对的峰面积,A2为样品前处理后基质加标测得的目标物定量离子对的峰面积。结果如图2所示,采用10 mL甲苯-正戊烷(1∶4)进行提取时的提取效率最高。
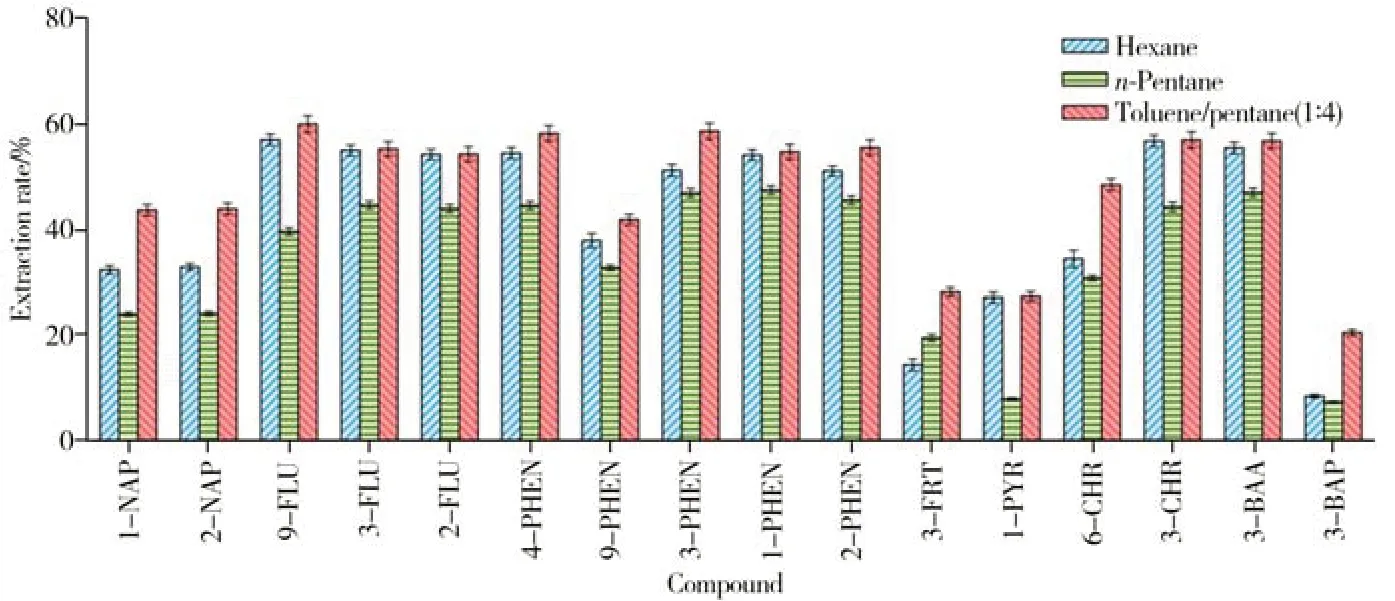
图2 提取溶剂对16种OH-PAHs提取效率的影响(n=6)Fig.2 Effect of extraction solvent on extraction rate of 16 OH-PAHs(n=6)
2.5 基质效应
人尿样品基质复杂,液液萃取过程中除目标物外,部分杂质会同时被提取。本研究使用硝酸银溶液进行除杂,以减少仪器分析时杂质对色谱柱效和质谱离子化效率的影响,降低基质效应的干扰。为评估基质效应对检测结果的影响,本研究使用同位素标记物作为定量内标,13C12-PCB105 作为回收内标(RS),分别评估了外标法和内标法的基质效应[1],其中外标法基质效应通过公式ME-ES(%)=(1-Aem/Aeo)×100%进行评估,式中Aem为基质中目标物与RS 峰面积比,Aeo为溶剂中目标物与RS 峰面积比;内标法基质效应通过公式ME-IS(%)=(1-Aim/Aio)×100%进行评估,其中Aim为基质中目标物与内标峰面积比,Aio为溶剂中目标物与内标峰面积比。当|ME| < 20%时说明基质效应较弱,不影响检测结果准确性。如表2 所示,所有目标物的ME-ES 范围为-7.2%~-37.7%,ME-IS 范围为-3.3%~-11.1%。表明因为同位素内标物与目标物具有相似的理化性质,本研究采用内标法定量可有效补偿基质效应的干扰。
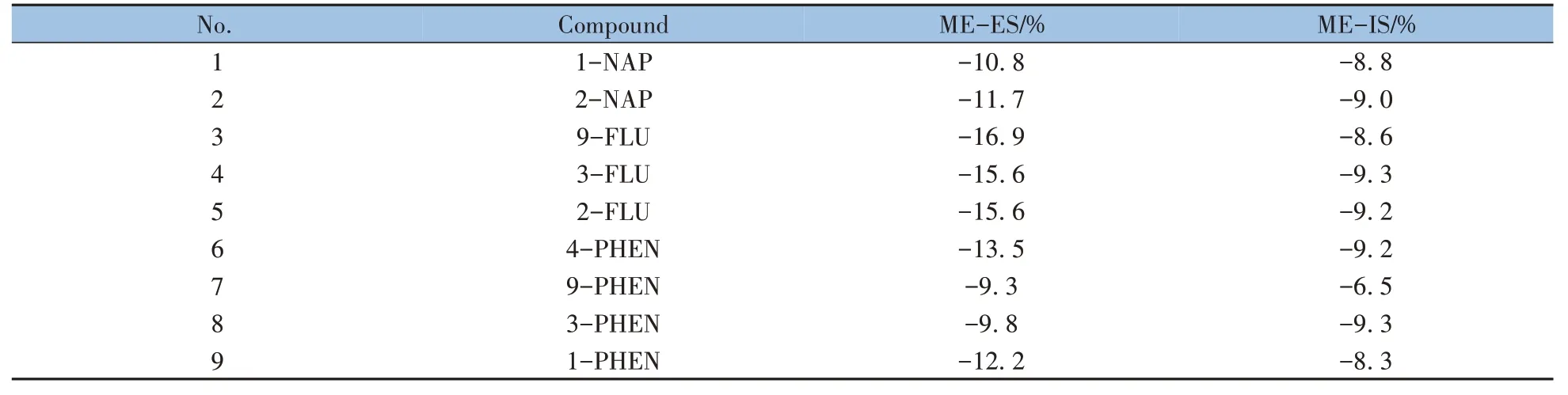
表2 16种OH-PAHs的基质效应Table 2 Matrix effects of 16 OH-PAHs
2.6 方法检出限与定量下限
采用甲苯配制目标物质量浓度分别为0.125、0.625、1.25、6.25、12.5、62.5、125 μg/L(1-NAP和2-NAP 为此质量浓度的5倍),定量内标质量浓度为6.25 μg/L,回收内标质量浓度为25 μg/L 的标准系列溶液。该标准系列中目标物换算为人尿中的质量浓度分别为0.02、0.1、0.2、1、2、10、20 μg/L(1-MAP 和2-HAF 为此质量浓度的5倍)。按照“1.4.3”衍生化后使用“1.5”方法进行测定,以目标物峰面积和对应定量内标峰面积的比值为纵坐标,其浓度的比值为横坐标,绘制标准曲线。如表3 所示,所有目标物在0.02~ 20 μg/L(1-NAP 和2-NAP 为0.1~100 μg/L)范围内线性关系良好,相关系数(r2)均大于0.99。
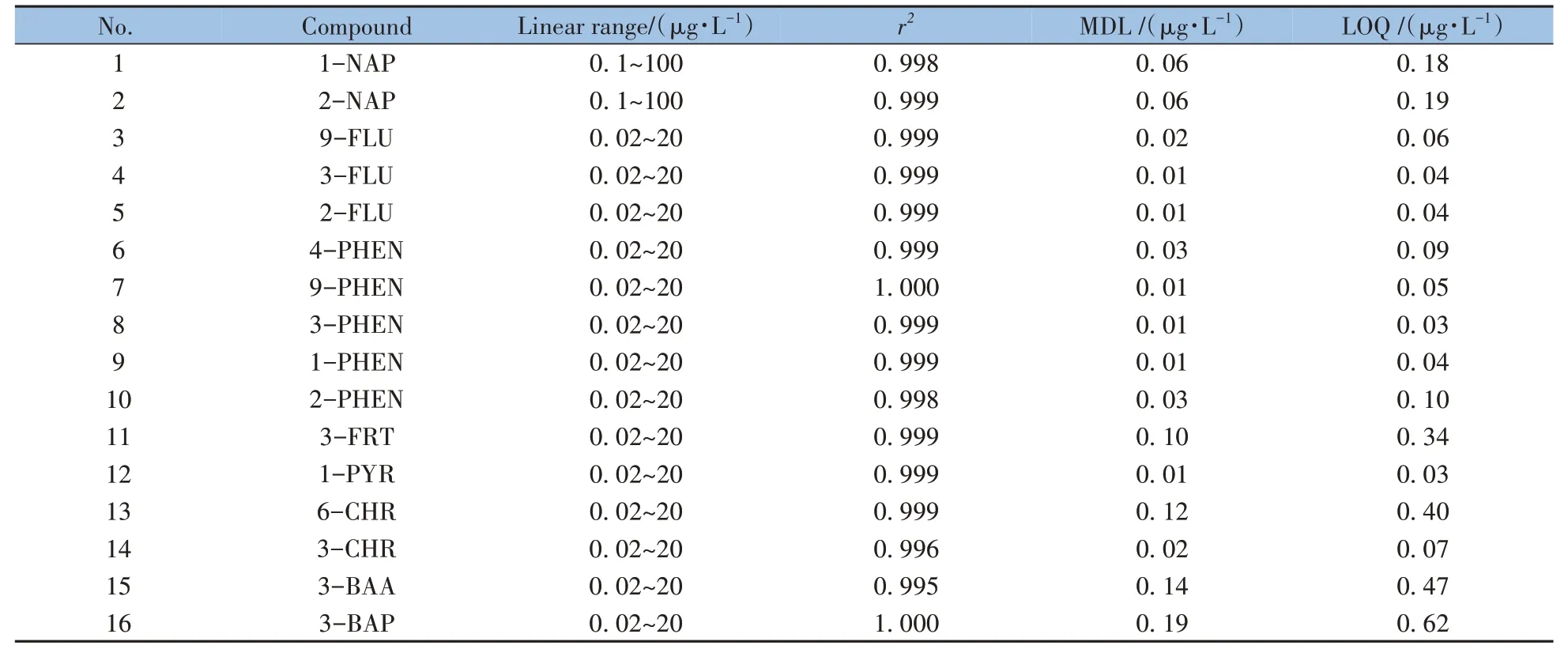
表3 16种OH-PAHs的线性范围、相关系数(r2)、方法检出限及定量下限Table 3 Linear ranges,correlation coefficients(r2),method detection limits(MDLs),and limits of quantitations(LOQs) of 16 OH-PAHs
根据“1.5”方法测定标准溶液的3 倍信噪比预估方法检出限。重复测定7 次浓度为5 倍方法检出限的基质加标样品,计算测定结果的标准偏差,方法检出限为3 倍标准偏差,方法定量下限为10 倍标准偏差[25]。如表3所示,本方法检出限为0.01~ 0.19 μg/L,定量下限为0.03~ 0.62 μg/L。
2.7 回收率与相对标准偏差
采用基质加标制备3个不同质量浓度水平的样品,同一天内重复测定6次评估方法回收率和日内相对标准偏差(RSD),连续6天每天各测定一次不同浓度水平的样品,评估日间RSD。如表4所示,16种OH-PAHs的加标回收率为81.6%~122%,日内RSD为0.22%~5.5%,日间RSD为0.76%~10%。
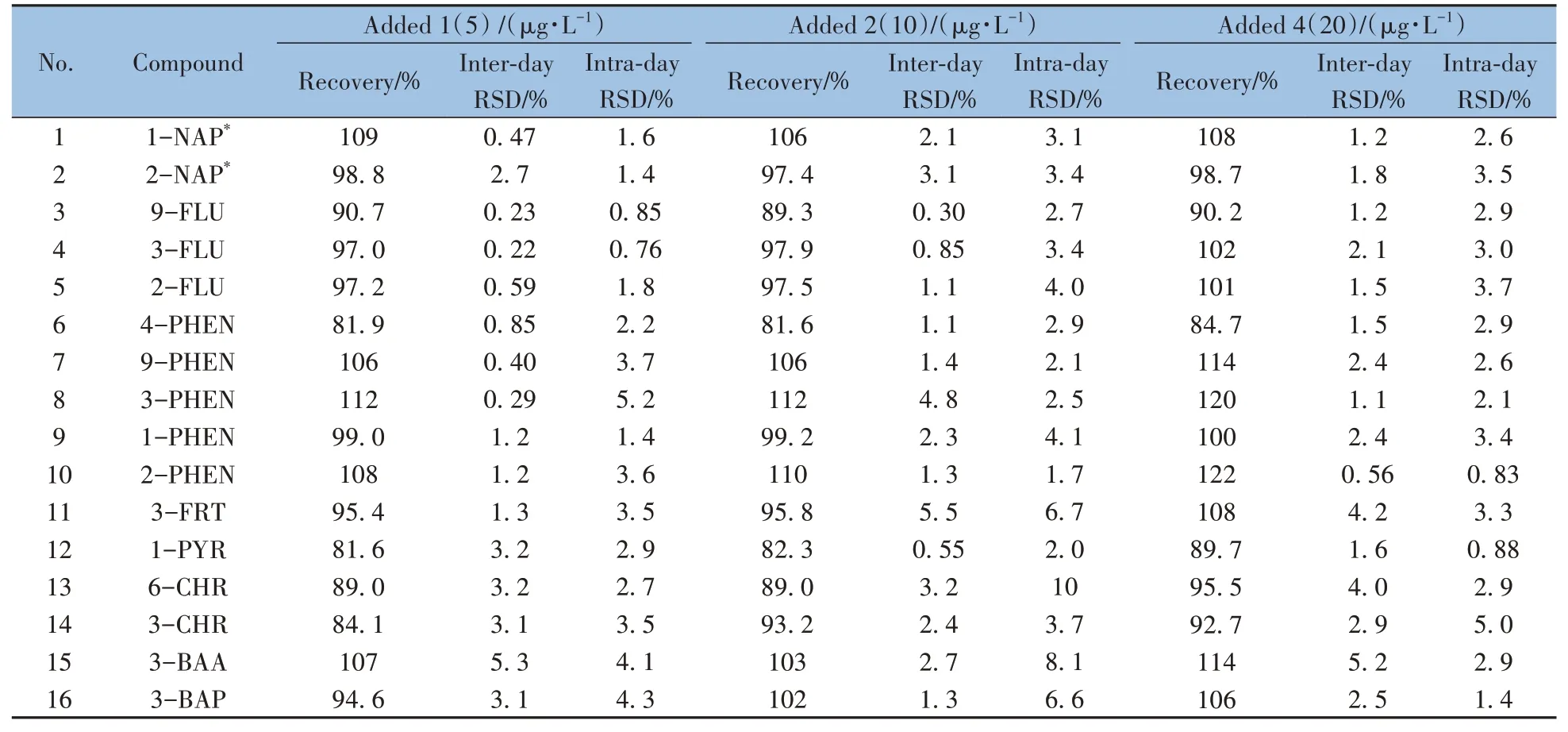
表4 16种OH-PAHs 的加标回收率和相对标准偏差(n=6)Table 4 Recoveries and RSDs of the 16 OH-PAHs(n=6)
2.8 标准参考样品测定
美国国家标准与技术研究院(National Institute of Standards and Technology,NIST)的SRM 3672 标准参考样品可用于评估10 种OH-PAHs 测定的准确度。结果显示,本方法的测得值与参考值的相对偏差均小于10%(表5),说明本方法的检测结果准确可靠。
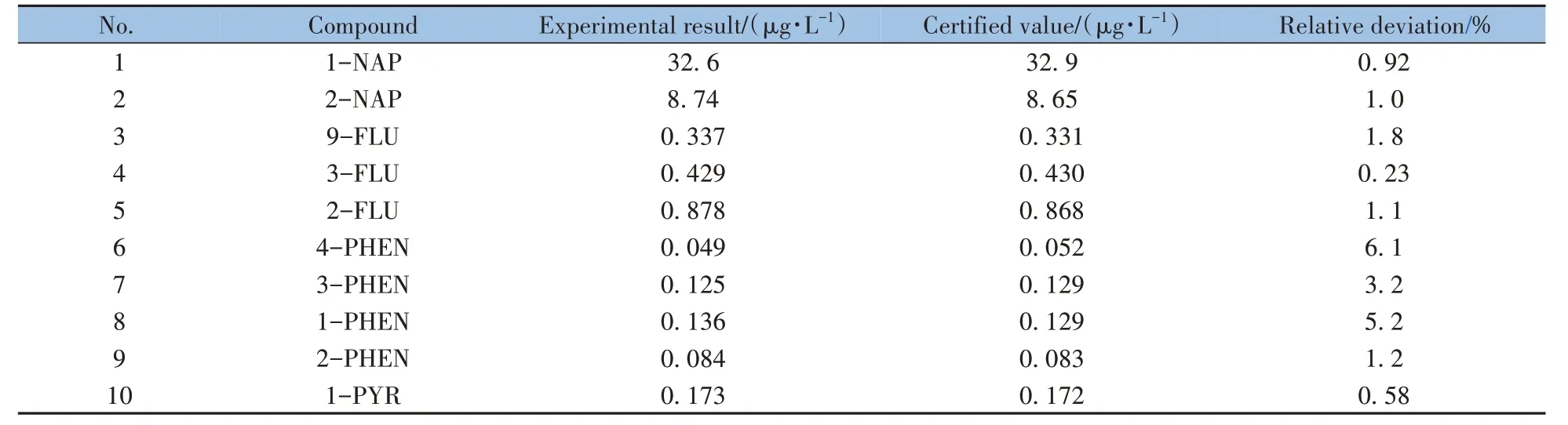
表5 标准参考样品(SRM 3672)的测定结果Table 5 Detection results of standard reference material(SRM3672)
2.9 实际样品测定
本方法应用于国家人体生物监测项目中,对采集的529份尿样进行检测,结果如表6所示。结果显示除3-CHR 外,其他15种OH-PAHs均有检出,其中1-NAP、2-NAP、9-FLU、2-FLU、1-PHEN、2-PHEN、3-FRT 和1-PYR 的检出率均超过90%;1-NAP 和2-NAP 的质量浓度水平高于其他组分,中位数(P50)分别为1.09 μg/L 和1.95 μg/L。该监测结果可为后续开展PAH 暴露对人群健康效应影响的研究提供数据支撑。
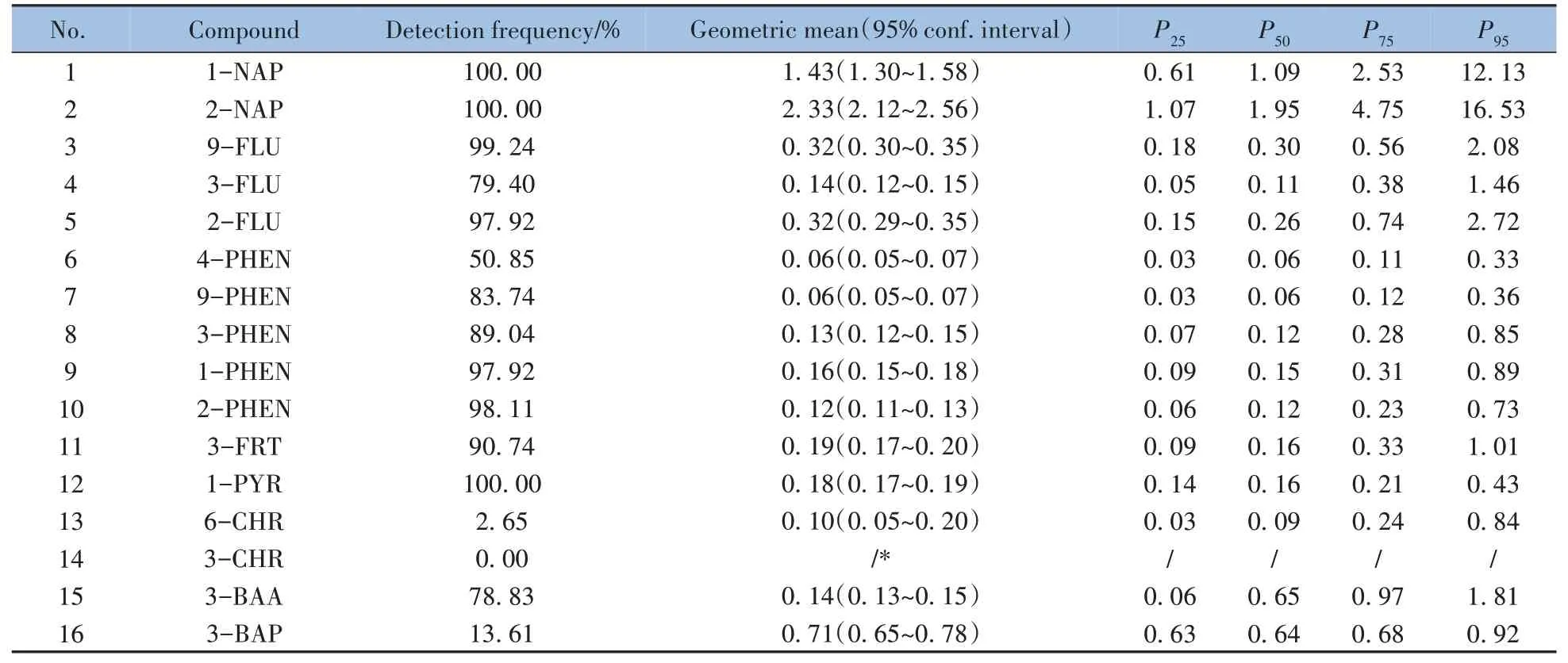
表6 尿样中16种OH-PAHs的检测结果Table 6 Detection results of 16 OH-PAHs in human unire samples
3 结 论
本研究基于LLE 的前处理方法,通过对实验条件的优化选择,建立了GC-MS/MS 同时测定人尿中16 种OH-PAHs 的分析方法。该方法检测成本低、分离度好、灵敏度高、基质效应干扰小,可为监测人群尿中OH-PAHs负荷水平和变化趋势提供技术支撑。
