基于UPLC-Q-Exactive MS 构建麻痹性贝类毒素的特征指纹溯源技术*
2024-02-24郑关超王潇潇翟毓秀谭志军吴海燕
张 帆 郑关超 王潇潇 翟毓秀 谭志军 吴海燕①
(1. 农业部水产品质量安全检测与评价重点实验室 中国水产科学研究院黄海水产研究所 山东青岛 266071; 2. 中国海洋大学食品科学与工程学院 山东青岛 266003; 3. 海水养殖生物育种与可持续产出全国重点实验室 中国水产科学研究院黄海水产研究所 山东青岛 266071)
麻痹性贝类毒素(paralytic shellfish toxins, PSTs)主要由Alexandrium、Gymnodinium和Pyrodinium等海洋甲藻产生, 通过贝类滤食进入食物链, 在世界范围内造成严重生态和食品安全风险, 是国际社会重点关注目标和管控对象(Viscianoetal, 2016; Nicolasetal, 2017)。研究表明PSTs 分布具有地域性特点(Villalobosetal, 2019), 而其风险形成过程主要受两个方面的影响: 首先是产毒藻的种类及来源差异, 链状亚历山大藻(Alexandriumcatenella)主要分布在渤海、黄海和东海海域(顾海峰等, 2011; 唐莹莹等,2018), 而太平洋亚历山大藻主要分布在东海和南海,链状裸甲藻在福建近海曾多次导致中毒事件(于仁成等, 2020)。其次, 不同地区贝类如紫贻贝摄食产毒藻后毒素成分也有显著差异, 在2019 年大连大窑湾海域紫贻贝中PSTs 成分以C1&2 和GTX2&3 为主(许道艳等, 2014), 而在浙南海域的紫贻贝中PSTs 成分则以C2、GTX5 和NEO 为主(张树刚等, 2011), 这种区域差异在其他研究中也得到了充分的证明(Yaoetal,2019; Liangetal, 2022)。因此, 贝类PSTs 组分的区域差异对其风险具有一定的指示作用, 可有效指向风险源地并用于产地溯源, 对于提升贝类中PSTs 风险的监管效果具有重要作用。
近年来, 指纹溯源技术广泛应用于生物地球化学、医学领域、环境监测、食品安全等多个领域, 通过对生物体指纹生物标志物的筛选, 结合数据库比对可快速实现源头识别(刘静等, 2022; 冷雪等,2023)。该技术是基于代谢组学技术, 以高通量、高灵敏度、高分辨率的现代仪器分析方法为手段, 对细胞、体液和组织中全部代谢物进行定性与定量分析,随后结合多元统计分析来构建指纹溯源技术模型(孙晓珊等, 2021; 高淑芳等, 2022)。其中液相色谱-质谱联用(LC-MS)(徐天润等, 2020)能够在任何单一分析平台中实现最高的代谢组覆盖率, 并且需要最少的样品前处理, 包括蛋白质沉淀和代谢物提取(Bujaket al, 2015), 适宜的提取溶剂、提取时间和处理温度等条件是影响前处理的重要参考指标。有文献报道, 基于LC-MS 的代谢指纹识别鲤鱼和虹鳟鱼精浆中的代谢物, 筛选出精子质量的新型生物标志物, 为优化人工繁殖奠定了基础(Dietrichetal, 2019)。同时也有研究针对澳大利亚翡翠贻贝新西兰绿唇贻贝和进行代谢指纹检测, 发现两种贻贝之间存在明显的代谢差异, 根据差异代谢物可以实现贻贝产地溯源(Rochfortetal, 2013)。此外, 也有研究采用同位素标记(Zhaoet al, 2019)、脂质(Shinetal, 2008)、微量元素(Morrisonetal, 2019)等技术识别双壳贝类肠道内容物的方式,实现贝类产地溯源(Gaoetal, 2006)。
采用高通量筛查技术获得的数据规模大且来源复杂, 因此需要结合化学计量学方法, 降低数据维度筛选出具有识别能力的变量(徐天润, 2020)。多变量分析是通过降低维度来简化数据的复杂程度, 并通过相关软件进行结果可视化, 主要包括主成分分析(principal component analysis, PCA)、正交偏最小二乘判别分析(orthogonal signal correction partial least squares-discriminant analysis, OPLS-DA)、聚类分析等。其中PCA 主要通过质量控制样本(QC)在PCA 图中分布情况对数据进行初步考察, 谭芷晴等(2023)为筛选炔诺酮暴露斑马鱼后的生物标志物, 运用PCA分析发现处理组与代谢组有明显分离。刘天亮等(2022)运用PCA 分析成功将来自山东、河南和河北产区的金银花样品分成了3 类。而OPLS-DA 可以使分类信息主要集中在主成分上, 并且通过变量投影重要度(Variable importance in projection, VIP)来确定差异化合物, 判别效果更具有说服力(李思源等, 2021)。然而, 基于多变量分析的代谢指纹从未用于研究摄食麻痹性贝类毒素的紫贻贝。
本研究选择我国近海主要麻痹性贝类毒素产毒藻种, 通过室内暴露贻贝模拟指纹信息传递过程, 采用UPLC-Q-Exactive MS 对五种前处理方法进行比较分析, 结合PCA 和OPLS-DA 化学计量学方法, 挖掘摄食不同产毒藻后贻贝体内的化学差异, 筛选区分摄食不同产毒藻贻贝中的特征指纹物质, 构建特征指纹溯源技术模型, 快速精准的识别潜在风险以便提高麻痹性贝类毒素风险源头识别。
1 材料与方法
1.1 实验材料
实验所用藻株包括链状亚历山大藻(A.catenella,GY-H25)、微小亚历山大藻(A.minutum, GY-H46)和链状裸甲藻(G.catenatum, GY-H65)购自上海光语生物科技有限公司。实验室内以f/2 培养液单种培养, 温度为(20±1) °C, 光照为6 000 lx; 光暗比12 h∶12 h。饵料藻选用小球藻(Chlorellavulgaris), 置于-20 °C冰箱保存。
样品前处理及色谱质谱条件优化采用取自秦皇岛贻贝重点养殖区的紫贻贝(Mytilusgalloprovincialis),经检测为麻痹性贝类毒素阳性贝, 毒素含量为1 008 µg STXeq/kg。
指纹溯源技术建立采用购于山东青岛码头紫贻贝(Mytilusgalloprovincialis), 平均规格为: 壳长(40.0±1.9) mm, 壳宽(22.6±0.8) mm, 总重(6.0±0.6) g,购入后先在实验室进行驯化, 每天持续通气并更换海水。实验共设置3 组, 每200 只紫贻贝为一组, 培养于24 h 通气的天然海水养殖容器环境下。
1.2 实验方法
1.2.1 紫贻贝室内暴露实验 各取1 mL 混匀后的链状亚历山大藻、微小亚历山大藻和链状裸甲藻的藻液, 加入20 μL 鲁哥试剂(Lugol’s agent)固定, 在显微镜下计数藻细胞密度。按照A.catenella组和A.minutum组投喂量约为1×105cells/(inds.·d),G.catenatum组投喂量约为 2×105cells/(inds.·d), 早晚各喂一次,轻微曝气。实验共持续14 d, 投喂产毒藻7 d, 投喂饵料藻7d 。分别取0 (对照)、1、3、7 和14 d 的紫贻贝样品, 每组样品设置3 个生物学重复, 毒素分析样品为3 只贝混样; 特征物质分析样品为6 只贝混样, 均解剖紫贻贝并采集全部软组织, 于-80 °C 下冷冻保存。
1.2.2 产毒藻和贻贝中麻痹性贝类毒素组成分析 将指数生长期的藻液取20 mL 于离心管中, 2 760 ×g离心5 min, 收集藻细胞置于15 mL 离心管中, 加入5 mL 1%乙酸水溶液。将其置于冰浴中, 使用超声波细胞破碎仪(JY92- ⅡN, 宁波新芝生物科技股份有限公司)对藻类样品进行破碎(设置全程时间5 min、间隔时间2 s、功率比50%)。镜检确认藻细胞完全破碎后, 将离心管置于高速冷冻离心机(Himac CR 22G,日本 Hitachi 公司)中, 2 760 ×g离心5 min, 供测试。将贻贝开壳用生理盐水清洗后取软组织, 沥水均质, 参考Boundy 等(2015)等方法进行麻痹性贝类毒素提取及测试。
1.2.3 特征指纹物质的前处理方法 取(1.00±0.02) g麻痹性贝类毒素阳性紫贻贝样品转移至5 mL 离心管(分为5 组, 每组6 个平行), 按照表1 中五种方法进行前处理, 提取液经12 470 ×g离心5 min 后, 供测试。
1.2.4 液相条件 Q-Exactive 液相色谱质谱联用仪(Q-Exactive, 美国 Thermo 公司)C8 色谱柱体系:Kinetex C8 (150 mm×2.1 mm, 2.6 µm); 梯度洗脱程序:0~0.5 min, 20% A; 0.5~5.0 min, 20%~90% A;5.0~14.0 min, 90% A; 14.0~15.0 min, 80% A。Amide色谱柱体系: TSK-Amide-80 柱(2 mm×15 cm, 3 μm);梯度洗脱程序: 0~1.0 min, 80% A; 1.0~4.0 min, 80%~40% A; 4.0~6.5 min, 40% A; 6.5~8.0 min, 40%~80%A;8.1~10.0 min, 80% A。
柱温: 40 °C; 进样量: 10 μL; 流速: 0.35 mL/min;流动相为: A 相为95%乙腈水(含2 mmol/L 甲酸铵,50 mmol/L 甲酸), B 相为水(含2 mmol/L 甲酸铵,50 mmol/L 甲酸)。
1.2.5 质谱条件 Q-Exactive 液相色谱质谱联用仪使用加热电喷雾离子源(HESI), 扫描模式: 全扫描/数据依赖二级子离子扫描(Full MS/dd MS2); 喷雾电压: 3 500 V; 毛细管和加热器温度: 320 和50 °C; 鞘气和辅助气: 40 和10 arb; 分辨率用半峰高处的全峰宽(FWHM)表示, 一级全扫描分辨率: 70 000 FWHM;trap 最大容量(AGC target): 1×106; C-trap 最大注入时间: 150 ms, 数据依赖二级离子扫描分辨率: 17 500 FWHM; C-trap 最大容量(AGC target): 1×105; C-trap最大注入时间: 50 ms; 质谱扫描范围: 质荷比(m/z)100~600 和600~1 500。
1.3 数据处理
1.3.1 样品前处理及色谱质谱分析 按照时间顺序在色谱图中随机选取峰强度大于1×105的10 个离子峰, 分别计算每种方法6 个样品的峰强度均值, 计算每种方法贻贝样品中所测的10 个离子峰峰强度及其变异系数(Coefficient of variation, CV=标准差/均值)。5 种前处理方法所测峰强度以均值表示, 多组间比较采用随机区组的单因素方差分析, 若组间差异有统计学意义, 进一步的两两比较采用Bonferroni 检验。以P<0.05 为差异有统计学意义。
1.3.2 特征指纹物质分析 原始数据使用Compound Discoverer 3.1 软件进行峰检测、保留时间对齐等预处理。采用数据库(ChemSpider, mzCloud 等)从精确质量数、同位素组成及分布与预测分子式的吻合度、一级质谱/二级质谱碎片和数据库匹配等方面进行化合物初步鉴定, 其中设置参数如下: 保留时间最大漂移值0.2 min; 质量偏差<5×10-6; 信号强度偏差<30%;峰强度最小值度50 000, 同时设置基质空白去除, 不进行统计学分析。由于特征物质没有标准品, 采用STX 标准品曲线进行相对定量分析计算其浓度。
1.4 统计分析
采用Excel 2021、Origin 2023、SIMCA 14.1 和IBM SPSS Statistics 22 软件对数据进行统计分析和绘图。在Excel 2021 中将数据整理为二维矩阵形式, 保留P值、m/z值、保留时间、分子式、峰面积等信息,根据P值筛选出差异显著的化合物(P<0.01); 以化合物的峰面积为变量,m/z值作为观测ID 将数据导入SIMCA 14.1 进行PCA 与OPLS-DA 分析; 根据VIP>1作为特征指纹物质。采用逐步判别法对3 组样品的13 个复合指纹物质特征建立3 个群体的Fisher 线性判别函数。
2 结果与分析
2.1 样品前处理及色谱质谱条件选择
通过对比不同色谱和质谱条件结果如图1 所示,两种色谱柱均为方法D 检测到的化合物数量最多。采用C8 色谱柱时(图1a), 正离子模式m/z100~600 扫描范围下检测到的化合物数量最多为3 374 个, 负离子模式m/z100~600 扫描范围次之; 采用Amide 色谱柱时(图1b)负离子模式m/z100~600 的扫描范围下检测到的化合物最多为1 785 个, 正离子模式m/z600~1 500 扫描范围次之。因此方法D 检测到的化合物达到了6 811 个, 更加适合对特征物质的提取。
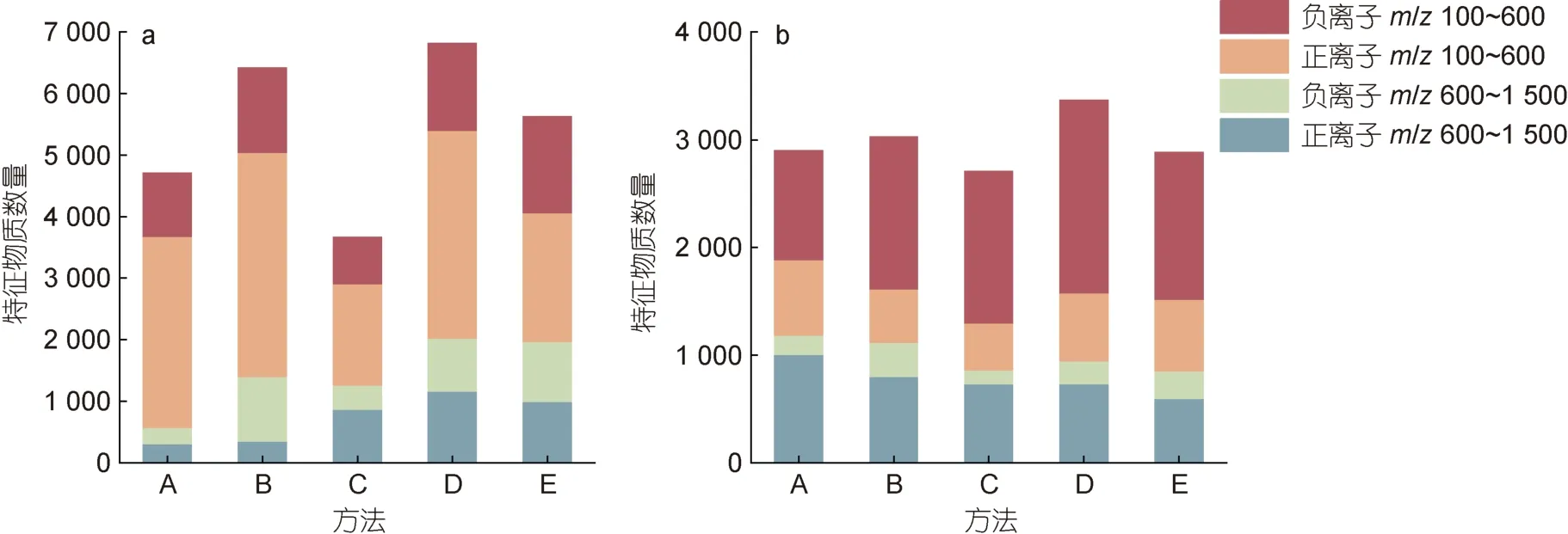
图1 不同前处理方法在每种色谱质谱条件下的特征物质数量Fig.1 The amount of compounds in each mode for different pretreatment method
此外, 根据不同方法的峰强度变异系数(CV 值)来分析不同的质谱条件。采用C8 色谱柱时, 离子峰强均值在正离子模式m/z100~600 和m/z600~1 500扫描范围下大于1×107且差异无统计学意义(P>0.05);比较其CV 值, 方法D 在正离子模式m/z600~1 500扫描范围下CV 值最小。采用Amide 色谱柱时, 方法D 在正离子模式m/z600~1 500 扫描范围下的峰强最高为5.49×107且差异具有统计学意义(P<0.05), CV 值<25% (表2)。因此, 选择方法D 在C8 色谱柱正离子模式m/z100~600 的扫描范围和Amide 色谱柱正离子模式m/z600~1 500 的扫描范围下更适用于贻贝的特征物质分析, 覆盖的化合物数量占总检测量的40.4%。在方法D 两种色谱质谱条件下, 分别在QC 样品色谱图的前、中、后随机共选取10 个离子峰进行分析, 其CV 值均≤1.00%, 各峰面积和峰强度的CV 值均<25.00%。以上结果表明本研究样品分析过程中仪器的精密度、稳定性及重复性均较好, 满足分析要求。

表2 不同前处理方法测得的峰强及CV 值Tab.2 Peak intensity and CV values of different pretreatment methods
2.2 紫贻贝摄食不同产毒藻后体内麻痹性贝类毒素分析
紫贻贝摄食不同产毒藻后麻痹性贝类毒素的变化情况如图2 所示, 在前7 天为毒素蓄积阶段, 紫贻贝中的麻痹性贝类毒素含量不断上升, 由暴露1 天毒素含量最低为54.1 µg STXeq/kg, 到7 天时毒素含量最高为5 538 µg STXeq/kg; 7 天到14 天为代谢阶段(停止投喂产毒藻), 至 14 天时毒素含量最低降至267 µg STXeq/kg。
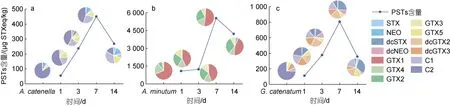
图2 紫贻贝摄食不同产毒藻后体内麻痹性贝类毒素含量及组成Fig.2 Variation of toxin content and composition after ingestion of different toxin producing algae by M. galloprovincialis
在投喂产毒藻的紫贻贝体内共检出11 种麻痹性贝类毒素成分, 不同组样品毒素成分差异显著。投喂A.catenella的紫贻贝中麻痹性贝类毒素主要成分为C1&2 和GTX5, 在整个实验中C1&2 占比从第1 天68.6%下降到第14 天的49.8%; 投喂A.minutum的紫贻贝中麻痹性贝类毒素主要成分为GTX1&4 和GTX2,且在整个实验期间各成分占比稳定; 投喂G.catenatum的紫贻贝中麻痹性贝类毒素主要成分为dcGTX2&3、dcSTX 和C1&2, 其中dcSTX 占比从第1 天的10.9%增高到第14 天的21.5%; 而C2 的占比从21.5%降至4.02%。因此, 摄食不同产毒藻后紫贻贝体内的毒素成分有明显差异且随时间发生代谢转化, 还需对紫贻贝体内的代谢物进行检测。
2.3 多元统计分析
2.3.1 主成分分析 本研究使用上述乙腈/甲醇/丙酮(体积比为1∶1∶1)作为提取剂的提取方法结合筛选出的两个离子通道, 经过软件处理后, 提取了3 886 个化合物, 利用P<0.01 筛选差异化合物并结合毒素成分进行PCA 分析, 如图3 所示, 前两个主成分分别解释了总变量的 36.9%和 18.3%, 共解释了55.2%的变量。三组样品分别沿PC1 或PC2 轴分离,表明摄食不同产毒藻贻贝中化合物存在差异。
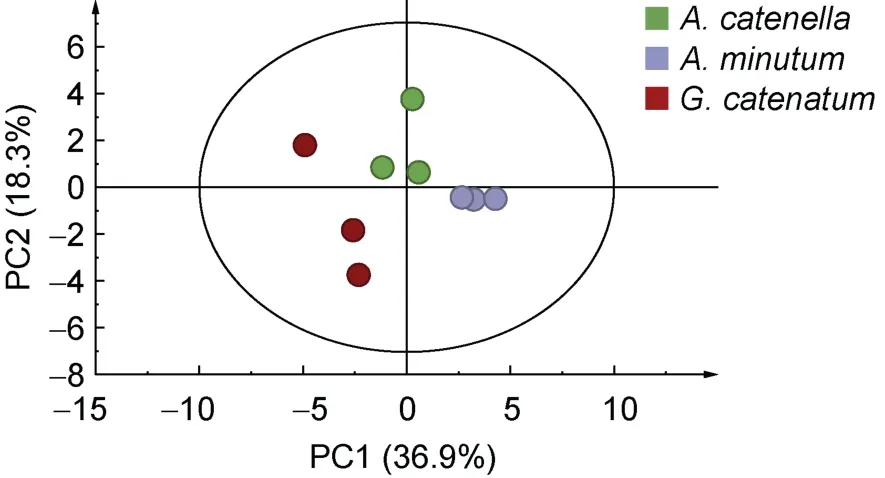
图3 投喂三种不同产毒藻后贻贝中差异化合物PCA 图Fig.3 PCA of different compounds and toxins in M.galloprovincialis fed with three different toxic microalgae
2.3.2 正交偏最小二乘判别分析 对投喂A.catenella、A.minutum和G.catenatum的贻贝经筛选后结合毒素进行OPLS-DA 分析, 如图4a~4c 所示。三组样品中的化合物显著分离, 自变量拟合指数分别为0.724、0.860、0.846, 自变量拟合指数分别为1.00、1.000、0.969, 模型预测指数(Q2)为0.985、0.982、0.988, 表明当前OPLS-DA 模型稳定可靠, 具有良好的预测性。经OPLS-DA 模型分析分别生成S-plots 图, 如图4d~4f 所示。OPLS- DA 模型中变量VIP>1.0 说明该变量对整体模型的贡献度高于平均水平, 作为本研究的毒素和特征指纹物质。此外, 毒素成分NEO 和dcNEO 作为摄食A.catenella和G.catenatum后紫贻贝的特有成分也被作为指纹物质,共计13 种复合指纹物质。根据精确分子质量、保留时间以及二级质谱碎片等与数据库进行比对参数如表3 所示。

表3 复合指纹物质Tab.3 Composite fingerprint substances
2.4 判别分析
根据筛选的复合指纹物质, 建立基于Fisher 判别函数的一般判别方法。以筛选出来的13 种复合指纹物质作为判别分析的自变量, 对贻贝样品进行多变量判别分析, 排除了7 种对分类作用较小的指纹物质,仅保留6 种指纹物质用于判别分析, 建立Fisher 线性判别函数如下:
式中:Y分别表示暴露于不同产毒藻贻贝的判别得分;C329.22、C381.14、C461.31、C374.21、CNEO、CdcNEO分别表示分子质量为329.22、381.14、461.31、374.21 的特征指纹物质和毒素NEO、dcNEO。回代检验正确判别率为100%, 交叉验证正确率为88.9%, 表明6 种特征指纹成分对贻贝的判别效果较好。
3 讨论
麻痹性贝类毒素的风险形成过程, 亦是从产毒藻信息向贝类传递的过程。通过传统的监管手段不仅耗费大量的人力物力财力, 对于近年来常见的小规模有害赤潮爆发作用有限(Fernandes-Salvadoretal,2021)。研究利用UPLC-Q-Exactive MS 结合PCA 分析与OPLS-DA 分析, 从摄食3 种麻痹性贝类毒素产毒藻的紫贻贝样品里共筛选出13 种复合指纹物质,对摄食不同藻的紫贻贝进行成功溯源。由于有毒藻种类和生态学特征不同, 其所产PSTs 的种类和含量也有很大差异(Guetal, 2013), 因此造成了贝类中PSTs风险表征差异明显, 这为识别PSTs 风险源头提供了重要依据。Lewis 等(2022)用苏格兰的链状亚历山大藻和英格兰南部的链状亚历山大藻分别投喂紫贻贝测定其体内的毒素谱, 根据毒素谱差异成功区分了摄食两种产毒藻的样品。同时有研究(Yanetal, 2022)表明不同产毒藻的毒素组成及毒性大小存在着较大差别, 并且我国的毒素分布也具有区域性特征。由于双壳贝类摄食产毒藻后毒素会在体内发生代谢转化,如摄食链状亚历山大藻后的紫贻贝中毒素成分随时间发生转化, C2 占比减小而GTX5 占比增加(张海涛等, 2023), 蛤仔在摄食微小亚历山大藻后毒素的积累阶段, GTX1&4 的占比逐渐增加, 在排出阶段占比下降( 焦玥 等, 2010), 这与本研究结果一致。在之前研究中发现这种代谢转化会受到投喂产毒藻量(Chenetal,2001)、环境的酸碱性(Cheetal, 2020)等因素影响, 因此仅根据毒素信息无法进行准确分类。
研究表明, 含水的有机提取剂如甲醇水溶液(Sidwicketal, 2017)可以有效地提取极性代谢物, 而非极性代谢物常选择氯仿、二氯甲烷等有机试剂(Caoetal,2020)进行提取。通过比较了5 种高效前处理方法, 经过扣除空白贻贝基质干扰后, 最终选择了乙腈/甲醇/丙酮作为提取剂的提取方法, 可以有效地移除大分子蛋白质(Lietal, 2017), 提高弱极性和非极性物质(Mushtaqetal, 2014)的提取效率。通过比较, 特征物质的提取方法和色谱质谱方法差异系数较小, 表明仪器稳定较好(李倩倩等, 2021), 可为后续特异性物质的筛选提供了方法。采用PCA 分析发现, 三组样品有明显分离; 进一步采用OPLS-DA 分析, 剔除无关数据使得模型更加易于解释, 可视化效果更加明显(钟若梅等, 2018)。刘鸽等(2021)采用PCA 与OPLSDA 分析, 通过VIP 值>1 以及P<0.05 筛选出暴露无机砷后三疣梭子蟹的100 种差异代谢产物。Vera 等(2019)等采用主成分分析(PCA)并比较独立建模(SIMCA)和偏最小二乘判别分析(PLS-DA)作为化学计量学分类方法, 实现了对品种特级初榨橄榄油的地理来源高灵敏和无偏差的鉴定。进一步采用Fisher线性判别模型构建判别函数, 可在变量服从正态分布、没有显著相关或变量的平均值和方差不相关使效果较好(李洪成等, 2017)。欧阳建等(2022)通过对黄金茶绿茶风味的测定将52 个样品分为醇厚型、鲜爽型和其他型3 种滋味类型。开建荣等(2022)构建枸杞原产地鉴别的判别模型, 模型的正确判别率达到了97.3%。目前, 未见Fisher 线性判别模型用于对摄食麻痹性贝类毒素产毒藻的溯源报道。本研究引入6 种物质构建的判别模型交叉验证准确率达到了88.9%,预测效果较为理想。指纹识别技术需要结合识别和鉴定(Cuadros-Rodríguezetal, 2021)两个过程, 为了达到更好地溯源目的还需要进一步的现场验证。同时,由于在同一海域往往会存在多种产毒藻且毒素成分会更加复杂, 如辽东湾曾检测出安氏亚历山大藻、伪亚历山大藻和李氏亚历山大藻等(宋伦等, 2018), 长江口海域则为塔玛亚历山大藻和链状亚历山大藻(高岩等, 2016)。因此, 需持续采集典型藻株特征物质识别以增加溯源技术的准确度和适用性。
4 结论
通过优化前处理方法, 确定了甲醇/乙腈/丙酮(体积比为 1∶1∶1)作为提取剂; 采用 Q-Exactive Orbitrap 筛选 C8 色谱柱中正离子扫描模式m/z100~600 的扫描范围和Amide 色谱柱正离子扫描模式m/z600~1 500 的扫描范围, 可获得40.4%的指纹信息。通过多元统计分析技术筛选出摄食不同产毒藻贻贝的13 种复合指纹物质, 构建了判别模型交叉验证准确率达到了88.9%。研究成果有助于提高麻痹性贝类毒素区域风险源头的准确识别能力, 为防范食用安全风险提供了科学依据和技术支撑。
