Pb 系简单钙钛矿氧化物PbMO3(M=3d 过渡族金属)的高温高压制备及物性研究
2024-02-24于润泽
于润泽
(北京高压科学研究中心, 北京 100193)
钙钛矿是目前功能材料研究中最重要的结构载体之一,其化学式一般表示为ABX3(A 一般为碱金属、碱土金属、主族Pb/Bi 和La 系元素;X 一般为阴离子),如图1 所示。近几十年,凝聚态物理及材料领域最重要的研究体系基本都是以钙钛矿结构为载体,如高温超导[1]、压电[2]、铁电[3]、巨磁阻[4]、太阳能电池[5]等。钙钛矿结构材料之所以有如此广泛的应用前景,主要是因为A 位、B 位以及X 阴离子位可以容纳不同种类的离子,将元素周期表中的元素排列组合可产生成千上万种化合物,这为在钙钛矿体系中探索奇异物理性质和开发新型功能材料提供了诸多可能。

图1 钙钛矿化合物的晶体结构Fig.1 Crystal structure of perovskite compound
Pb/Bi 系列钙钛矿氧化物材料是该体系中的典型代表。作为主族元素的Pb/Bi 的最外层电子为6s2,价态可表现为Pb2+/4+或Bi3+/5+,即价态跳跃(valence-skipping)行为[6-7],在形成化合物时,相应出现了+2/+4 或+3/+5 价态共存现象,表现出了奇异的电荷序。由于6s2孤对电子的作用,Pb/Bi 系化合物很容易出现极化结构,表现为铁电极化等行为。进一步地,如果在B 位引入磁性离子,则可能出现极化结构和磁性共存,这为寻找磁电耦合材料提供了一种可能。此外,由于Pb 中的6s电子轨道非常接近3d轨道,随着M 元素原子序数的增加,3d轨道的能级逐渐加深,与O2p轨道的耦合也逐渐变化,导致PbMO3材料体系中的电荷价态、晶体结构和磁性等均发生系列演化,在外部条件的调控下会出现诸如结构相变、电子相变和电荷转移等奇异的物性[8]。
本文将基于作者和合作者近十余年的研究成果,同时结合国外相关研究组的工作,对Pb 基简单钙钛矿氧化物材料的高压制备和物性研究进行较为系统的梳理。Bi 系简单钙钛矿氧化物材料已有较为详细的综述[8],在此不再赘述。
1 PbVO3 的高压制备和物性研究
2004~2005 年,Shpanchenko 等[10]和Belik 等[11]陆续报道了高压合成PbVO3钙钛矿材料。该材料表现出与PbTiO3同构的四方晶体结构,如图2 所示。但是PbVO3钙钛矿材料的c/a轴比约为1.23,远大于PbTiO3的轴比1.06[12],表明PbVO3具有极大的晶体结构畸变,电子价态为Pb2+V4+O3,电阻测试表现出半导体行为。遗憾的是,由于其轴比较大、电阻率较低,实验上还无法观测到铁电行为,只能认为是一种热释电材料。Belik 等[11]同时发现在压力作用下这种材料会从四方结构相变为立方结构,并伴随着从半导体到金属的转变。单晶磁性测试发现,这种材料在180 K 表现出二维反铁磁行为,谬子自旋弛豫/旋转(μSR)测量发现该材料在43 K 时形成了反铁磁结构[13]。尽管在实验过程中没有发现PbVO3的铁电行为,但是近期通过对其进行电子掺杂以及与其他化合物互掺,在实验上观察到了负热膨胀[14]和准同型相界(morphotropic phase boundary,MPB)[15]等性质。
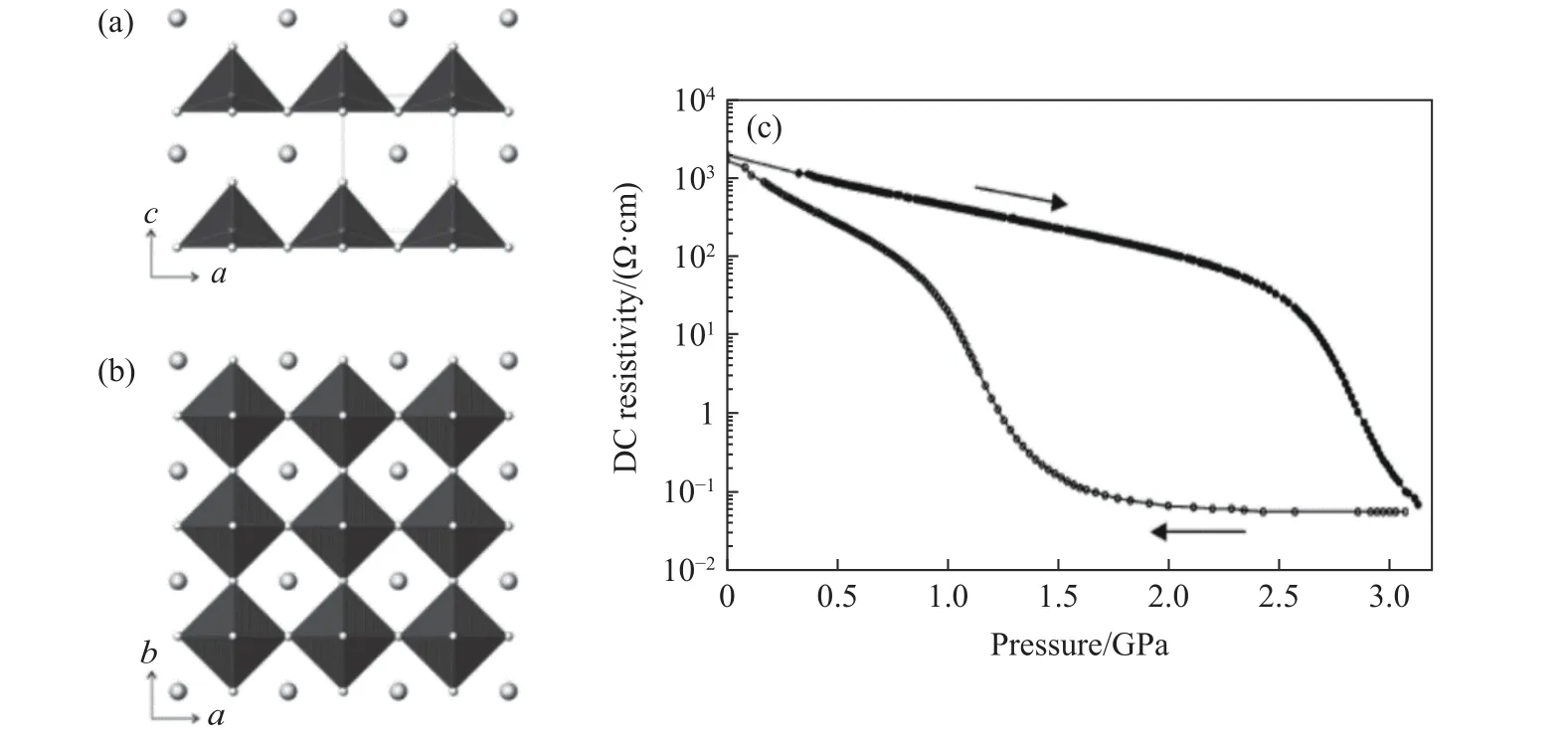
图2 PbVO3 的晶体结构[10-11]:(a)沿b 方向;(b)沿c 方向;(c) 直流电阻率-压力演化曲线Fig.2 Crystal structure of PbVO3[10-11]: (a) along b axis; (b) along c axis; (c) DC resistivity-pressure evolution curves
2 PbCrO3 的高压制备和物性研究
早在半个世纪前,科学家们就已经通过高压实验方法合成了PbCrO3化合物,因此PbCrO3并不是一种新材料[16-18]。早期的物性表征研究发现,PbCrO3呈现立方构型,是一种磁性为G型的反铁磁结构,总磁矩S=1,并据此推断出Cr 的价态为+4,电输运测试表现为半导体行为。在完成初步物性表征的同时,研究者还发现PbCrO3具有一些反常行为。首先,该材料的晶格常数呈反常增大。简单立方钙钛矿中的晶格常数主要由A 位和B 位的离子半径决定。在PbCrO3体系中,早期的磁性测试表明,Cr 的价态为+4,理论上讲Pb 的价态应该为+2。与其同构的SrCrO3相比,尽管在12 配位情况下A 位离子半径相近[9],但PbCrO3的晶格常数却比SrCrO3大4.8%。此外,早期研究发现,其X 射线衍射(X-ray diffraction,XRD)的衍射峰表现出了反常的宽化,在背底上还出现了许多“鼓包”(scattering),提示其中可能包含丰富的短程结构畸变。2010 年,Xiao 等[19]发现PbCrO3在高压下发生了等结构相变,并伴随约为10%的体积收缩,如图3所示,这种现象极为罕见。这些反常行为引起了研究者们的极大兴趣,从2007 年开始,国际上几个从事高压固态化学研究的课题组围绕着上述问题对该体系开展了一系列深入的研究工作。
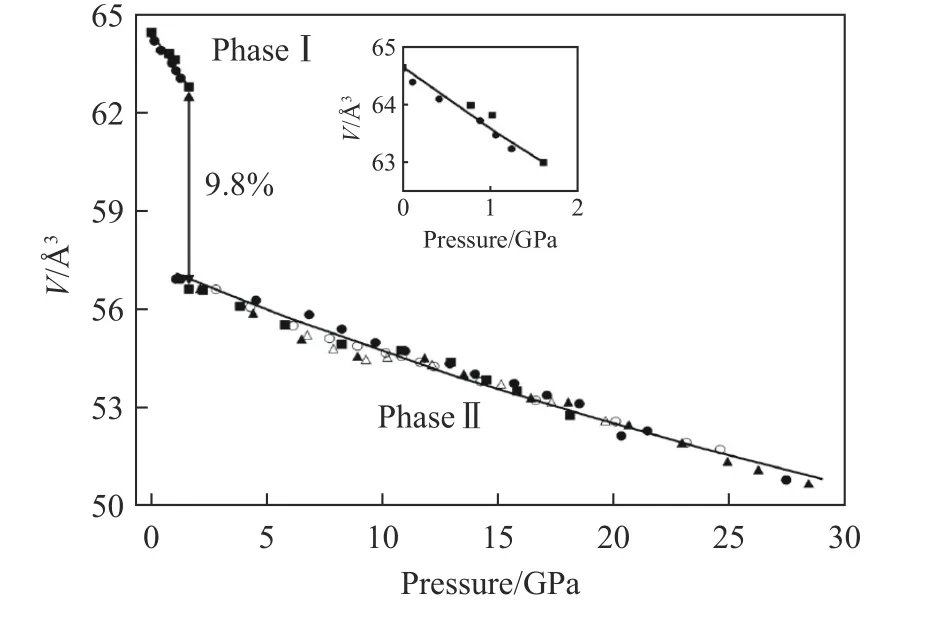
图3 高压下PbCrO3 的巨幅体积收缩[19]Fig.3 Large volume collapse of PbCrO3 under high pressure[19]
首先,西班牙马德里大学的Arévalo-López 等[20]利用高压技术制备了该材料,采用高分辨电子显微镜并结合能量散射型X 射线荧光光谱仪(EDX),发现材料中的Pb 位出现了缺位,并且晶体结构表现出远比简单立方结构复杂的微观结构,并据此认为其微观结构来源于Pb 的缺位。但是,他们的实验存在一个严重的问题,即样品的成分分析并不是基于纯相样品,同时他们也没有解释为什么这个体系会出现反常的晶格常数,以及巨幅压致体积收缩等行为。
2013 年,中国科学院高能物理研究所的Wu 等[21]做了一些新的尝试。如图4 所示,他们首次利用X 射线吸收谱研究了高压下Cr 元素的价态变化,发现常压下Cr 的价态应该为2Cr3++Cr6+,在高压下发生了Cr3+与Cr6+之间的电荷转移,并最终全部变成Cr4+,同时伴随半导体到金属的转变。他们提出,大幅体积收缩可能起源于价态的变化,该工作首次注意到了元素价态在其中的重要作用。为了进一步研究其本征物性,我们通过后处理酸洗法获得了高纯样品,并通过高分辨电子显微镜发现了复杂的微观结构,Pb-Pb 之间的距离分布在3.8~4.2 Å之间,同时发现纯相样品并不存在Pb 缺位,排除了复杂的微观结构来源于Pb 缺位的结论[22]。进一步地,Cheng 等[23]发现样品中存在2Cr3++Cr6+的电荷分布,在压力作用下发生了电荷转移,最后在高压下全部变成了Cr4+,同时伴随绝缘体到金属的相变以及巨幅体积收缩,至此给出了一个较为完美的解释。但是,这其中还存在一个问题,关于Pb 的确切价态,尤其是其在高压下的行为,还没有直接的实验证据。
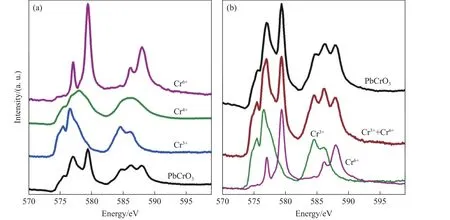
图4 (a) PbCrO3 中Cr-L2,3 的吸收边,(b) PbCrO3 中Cr 的X 射线吸收谱[21]Fig.4 (a) Cr- L2,3 edge in PbCrO3; (b) X-ray absorption spectra of Cr in PbCrO3[21]
近期,中国科学院物理研究所的Zhao 等[24]与德国马普所的研究者合作,利用高分辨X 射线吸收谱详细研究了PbCrO3材料中Pb 的电子结构以及高压下的演化行为。如图5 所示,他们通过测量发现,PbCrO3中A 位的Pb 离子呈现Pb2+单一价态,而B 位的Cr 离子由6 配位的Cr3+和4 配位的Cr6+按2∶1 构成,从而使得该材料呈现价态分布,进一步证实了Cheng 等[23]的研究结果,解释了PbCrO3在常压环境中呈现绝缘性的物理起源。再进一步的压力调控显示,随着压力增加,在绝缘体-金属化相变前后,PbCrO3中的A 位Pb 离子始终呈现Pb2+单一价态,如图6 所示。以上结果表明,PbCrO3材料中压力诱导的晶体结构和电子结构的急剧变化来自于压力抑制了Cr 离子的电荷歧化,导致2Cr3++Cr6+→3Cr4+电荷价态分布演化。该研究澄清了自PbCrO3材料发现以来的化合价态争议,揭示了其在常压/高压环境下绝缘性和金属化相变构效起源。经过半个世纪的研究,最终给这个极具争议的课题画上了完美的句号。在电子态研究的同时,围绕PbCrO3本征的物性研究也有了新的进展:如Wang 等[25-26]通过实验研究了PbCrO3的莫特特性和莫特临界点的巨大黏滞弹性;Han 等[27]首次在高压下获得了PbCrO3单晶样品。
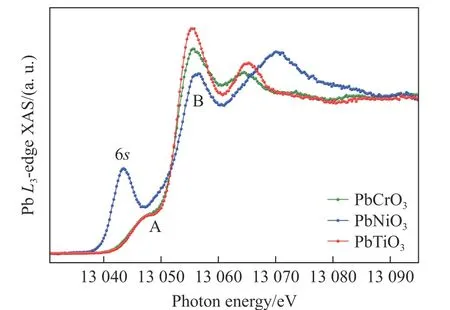
图5 PbCrO3 中高分辨X 射线吸收谱的Pb-L3 吸收边[24]Fig.5 Pb-L3 edge of high-resolution X-ray absorption spectra in PbCrO3[24]
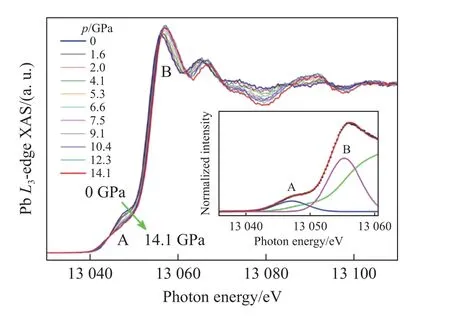
图6 PbCrO3 中高分辨X 射线吸收谱的Pb-L3 吸收边随压力的演化[24]Fig.6 Pressure dependent Pb-L3 edge of X-ray absorption spectra in PbCrO3[24]
3 PbMnO3 的高压制备和物性研究
2002 年,Bougerol 等[28]在7.8 GPa 和880 ℃条件下通过高压技术制备了Pb-Mn-O 三元钙钛矿化合物,分析得出其化学式为PbMnO2.75,晶体为六方钙钛矿(6H)结构,初步确定空间群为A2/m。考虑到六方钙钛矿晶体随着合成压力的增大会出现诸如2H、9R、4H、6H 以及3C(立方)等结构,Oka 等[29]进一步研究了不同合成压力下材料的晶体结构及物性。首先,在8 GPa、1 073 K 条件下制备了样品,分析其空间群为C2/c。在磁性测试中,155 K 附近出现了反铁磁结构,但是在低温下的零场冷(ZFC)和场冷(FC)磁性测试曲线并没有重合,表明其在低温下可能出现倾斜的弱铁磁性。进一步提高合成实验的压力,在15 GPa、1 273 ℃条件下,发现样品转变成了四方结构(P4/mmm),更为重要的是,在四方结构中c/a= 1.017,已经非常接近立方钙钛矿结构。磁性测试表明:其在20 K 左右出现了反铁磁转变,但是该温度明显低于很多具有Mn4+价态的化合物,如SrMnO3(233 K)[30]和CaMnO3(130 K)[31]。据此,Oka 等[29]认为这可能是由于样品中存在氧的缺位,出现了Mn3+所致。根据Goodenough-Kanamori 法则,eg轨道半满情况下容易出现铁磁转变,Mn3+具有电子态,而 Mn4+具有电子态,Mn3+和Mn4+的铁磁耦合打乱了Mn4+的反铁磁耦合,进而降低了磁转变温度。
此时,与样品相关的很多性质,如实验上能否观察到立方结构、Pb 和Mn 的电子态以及能否制备无氧缺位的样品等都还没有解决。近期,Li 等[32]为进一步深入研究这些问题,利用大腔体高温高压合成装置成功制备了PbMnO3钙钛矿材料。高分辨同步辐射X 射线粉末衍射和光学二次谐波结果表明,PbMnO3钙钛矿是空间群为P4/mmm的非极性四方结构而非立方结构,如图7 所示,这在容忍因子t>1的钙钛矿体系中极为罕见,同时通过实验证实了PbMnO3钙钛矿是一种非铁电材料。高分辨X 射线吸收谱结合热重分析显示,PbMnO3钙钛矿发生了电荷歧化,其电荷组态为如图8 所示。一方面,由于在上述同步辐射X 射线衍射图谱中并未发现超晶格衍射峰,并且结构精修结果表明,Pb 离子和Mn 离子均具有较大的热振动因子,因此,可以推断不同价态的Pb 离子和Mn 离子在晶格中无序排列;另一方面,由于Mn3+离子受其八面体配位晶体场的影响,产生的晶体场分裂能Δc将在一定程度上抑制从Pb 离子到Mn 离子的电荷转移,从而导致PbMnO3表现出奇特的电荷分布。
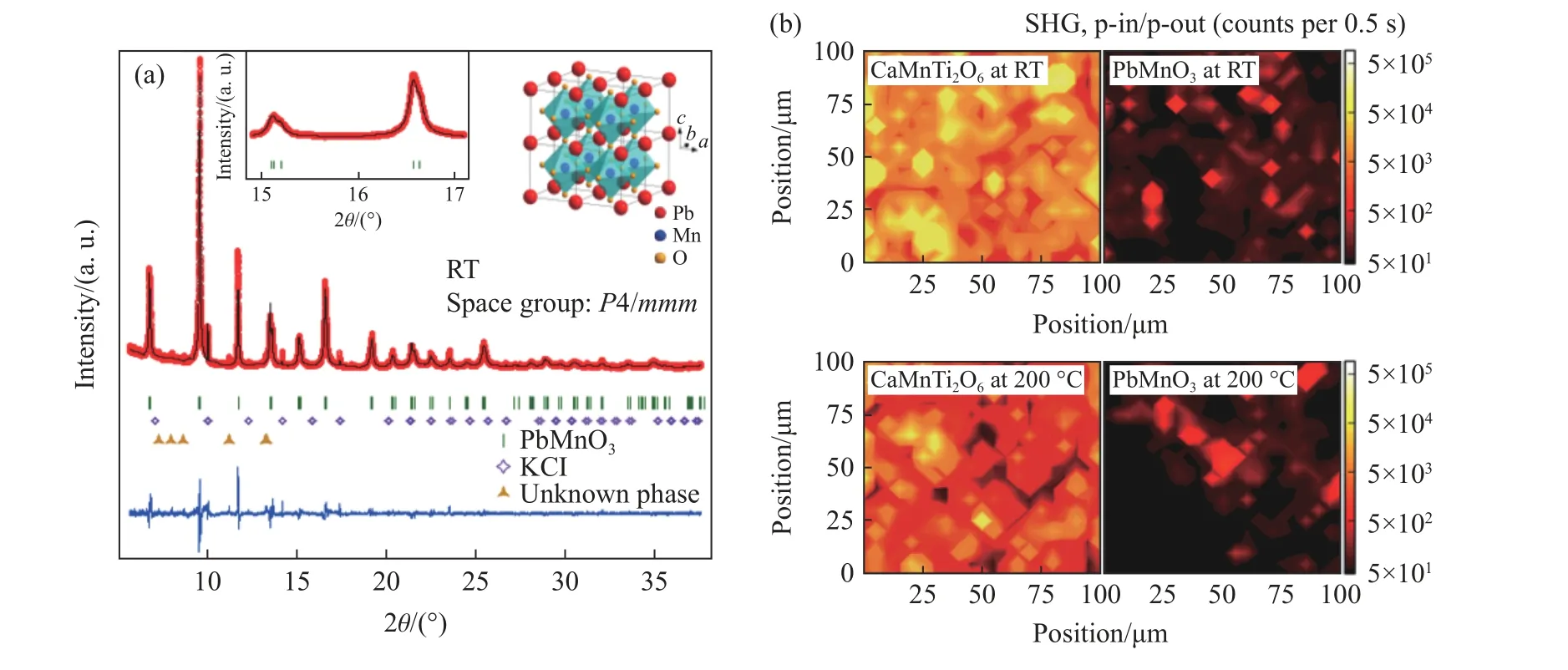
图7 PbMnO3 的(a)同步辐射X 射线粉末衍射谱图和(b)二次谐波光谱[32]Fig.7 (a) Synchrotron X-ray diffraction and (b) second harmonic generation of PbMnO3[32]
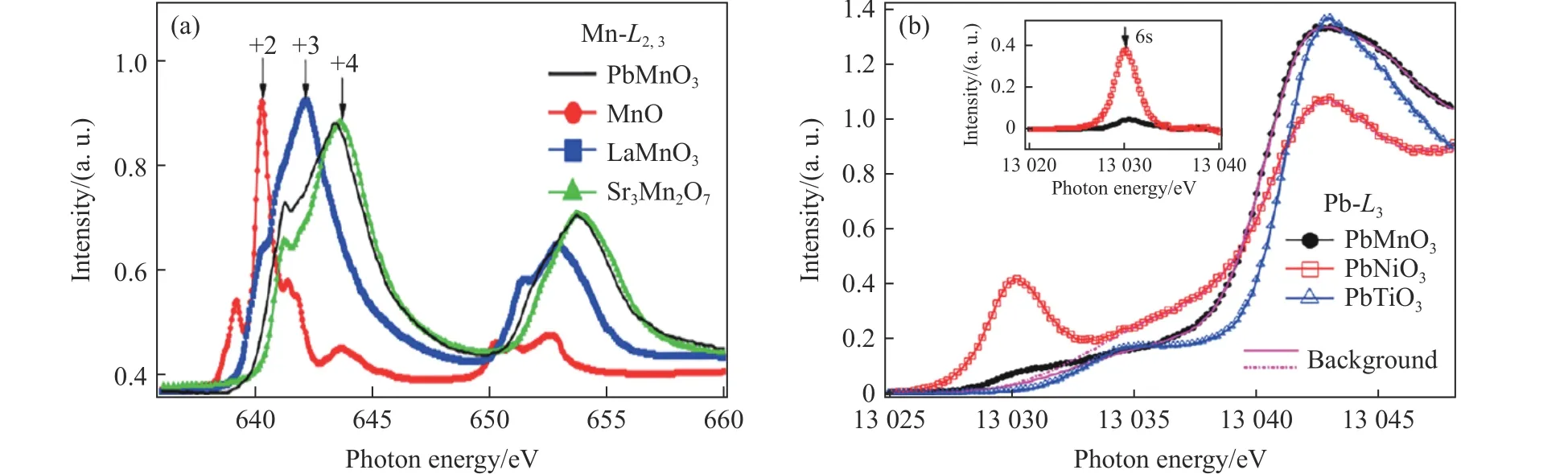
图8 PbMnO3 的X 射线吸收谱:(a) Mn-L2,3 吸收边,(b) Pb-L3 吸收边[32]Fig.8 X-ray absorption spectra in PbMnO3: (a) Mn-L2,3 edge, (b) Pb-L3 edge[32]
进一步的PbMnO3电磁输运特性测量结果表明:(1) 虽然其电阻率表现出绝缘体行为,但与晶界散射无关的室温热电势远小于普通绝缘体。(2) 磁化率显示其在0.1 T 低磁场作用下发生反铁磁转变,而在9.0 T 高磁场作用下发生铁磁转变。最为重要的是,Arrott 曲线的高场部分呈现良好的线性关系,且居里温度处的直线反向延长线经过原点,表明该材料具有巡游铁磁性。(3) 同样与晶界散射无关的比热测量证实,低温部分电子贡献较大,较宽的磁转变温区与磁化率结果吻合,如图9 所示,其中:S为热电势,c为比热容,M为磁化强度,H为磁场强度,χ 为磁化率。基于密度泛函理论第一性原理计算结果也表明,PbMnO3具有铁磁性金属基态。考虑到非本征的电阻率测量可能与晶界散射有关,并不能反映材料的本征物性,因此,可以说PbMnO3是PbMO3体系中唯一的铁磁性金属。
4 PbFeO3 的高压制备和物性研究
2006 年,日本学习院大学的Inaguma 教授首先报道了PbFeO3材料,且初步表征其晶体结构为正交结构,具有电子态[33]。但是,由于XRD 衍射峰极为复杂,很多衍射峰都重叠在一起,解析其晶体结构变得极为困难,因此,在接下来的十几年中一直没有进展。笔者早期在日本工作期间就注意到了该材料体系,受限于当时的实验条件一直没有做深入研究。最近,笔者联合中国科学院物理研究所以及日本、德国团队在该课题的研究中取得了突破性进展。
经过大量尝试探索,团队成员获得了制备高质量PbFeO3多晶样品的最佳条件,即合成压力必须高于8 GPa,加热温度为1 150 ℃(上下浮动不超过50 ℃)。高质量样品的合成为下一步的物性研究奠定了坚实的基础。为确定材料的晶体结构及综合物理性质,团队进行了同步辐射X 射线衍射、X 射线吸收谱、高分辨电子衍射、磁化率、磁化强度、电输运、变温中子衍射等系列实验测试以及基于第一性原理的理论计算。结果表明,PbFeO3的B 位由单一Fe3+离子组成,但A 位由Pb2+和Pb4+2 种电荷态按1∶1 比例组合而成。并且,Pb2+与Pb4+在A 位形成全新的—A—B—B—型层状电荷有序,其中:A 层为单一Pb2+,B 层为75%Pb4+和25%Pb2+有序组合而成。这种新颖的电荷有序在其他化合物中未曾报道,使得材料形成2ap×6ap×2ap的超晶胞(ap为简立方钙钛矿的晶格常数)。这相当于A 位为特殊的电荷有序,B 位Fe3+离子在A—B 与B—B 层间形成具有不同对称性的2 种原子位置,导致FeO6八面体形成不同的各向异性晶体场,如图10 所示。PbFeO3在600 K 发生倾斜反铁磁自旋有序,自旋取向平行于b轴,宏观上表现出弱铁磁性。令人意外的是,该材料在高达418 K 的临界温度时自旋取向发生90°旋转,随着温度降低形成自旋平行于a轴的共线反铁磁结构,如图11 所示。在以往的Fe 基氧化物中,自旋重取向往往发生在室温以下,且与A 位稀土离子或者B 位磁性离子掺杂引入的磁各向异性相关[34-36]。理论计算表明,PbFeO3钙钛矿中A 位特殊的层状电荷有序导致了B 位2 种Fe 位置磁各向异性的竞争,其竞争结果是自旋重取向转变的主要原因。由此可见,层状电荷有序是有效调控自旋取向的一种新机理,PbFeO3远高于室温的自旋重取向行为有利于该效应在自旋电子学器件领域的应用[37]。
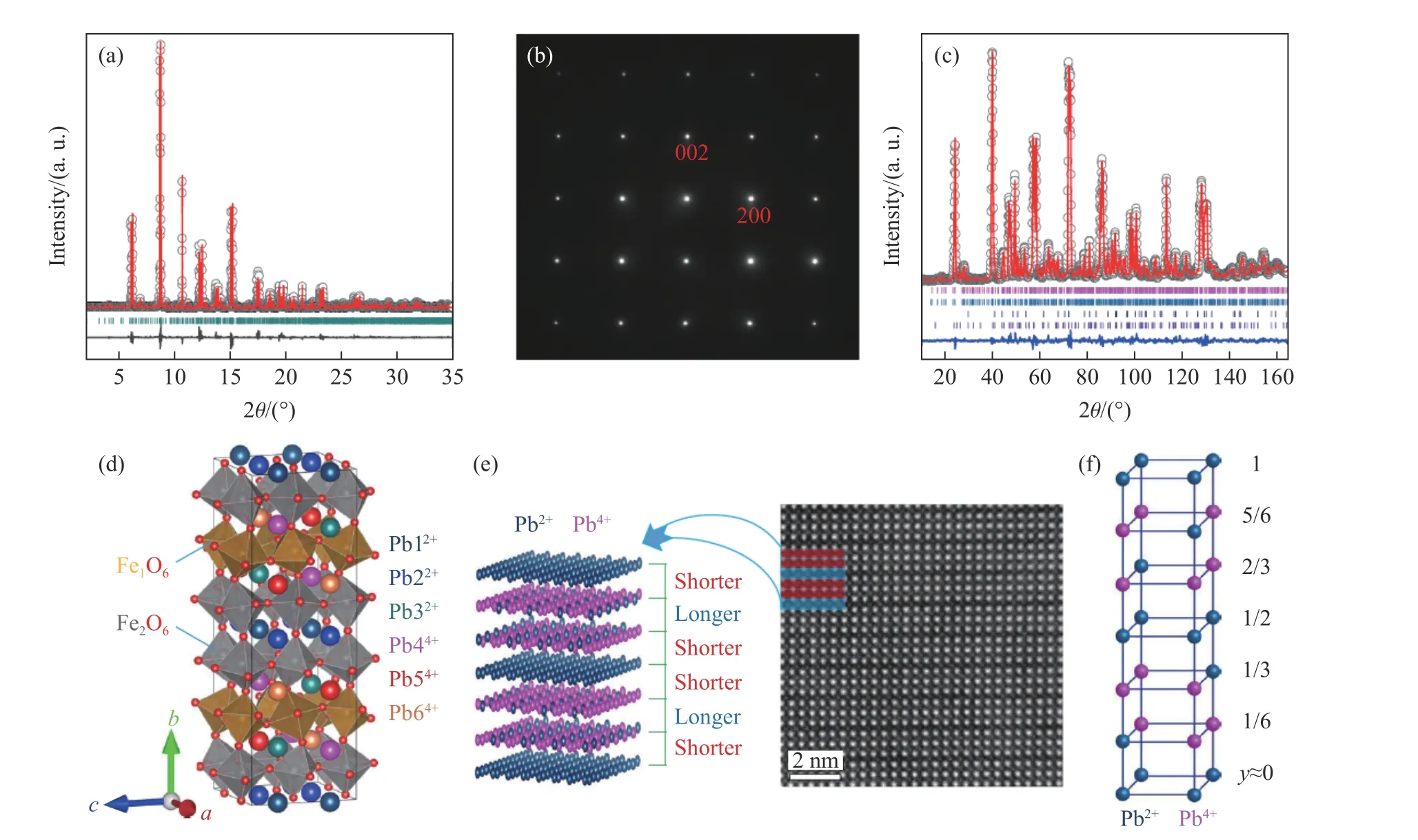
图10 PbFeO3 的 (a) 同步辐射X 射线衍射谱、 (b)电子选区衍射图、(c)中子衍射谱以及(d)~(f)晶体结构和电荷序[37]Fig.10 (a) Synchrotron X-ray diffraction pattern, (b) selected electron diffraction, (c) neutron diffraction,(d)-(f) crystal structure and charge order of PbFeO3[37]
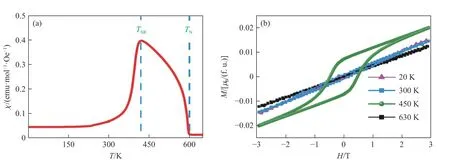
图11 PbFeO3 的(a)磁化率曲线、(b)磁滞回线、(c)~(d)温度诱导的自旋重排[37]Fig.11 (a) Magnetic susceptibility, (b) isothermal magnetization loops, (c)-(d) temperature induced spin reorientation of PbFeO3[37]
5 PbCoO3 的高压制备及物性研究
如果说PbFeO3材料是PbMO3材料研究中的最后一块拼图,那么PbCoO3则是倒数第2 块拼图。PbCoO3材料研究之所以迟迟没有进展,主要是由于其实验条件非常苛刻,需要极高的温度和压力,往往接近实验设备的极限。近期,中国科学院物理研究所的龙有文研究员团队与日本东京工业大学Azuma 教授团队合作,利用高温高压技术第一次成功制备了具有钙钛矿结构的PbCoO3体系,并发现了该物质奇特的电荷属性及其他物理性质[38]。实验表明,PbCoO3具有十分苛刻的成相条件,压力不能低于10 GPa,成相温度变化范围不超过50 K,这种敏感的合成条件可能预示着多种电荷态的竞争。他们对该样品进行了包括单晶X 射线衍射、中子衍射、电子衍射、X 射线吸收谱、磁化率、磁化强度、电阻、比热容以及理论计算等系列研究。结果表明,虽然此化合物具有简单的PbCoO3化学式,但A 位的Pb 具有1∶3 有序的Pb2+与Pb4+价态分布,而B 位的Co 具有1∶1 盐岩型有序的Co2+与Co3+价态分布,并且Co2+为有磁性的高自旋态(S=3/2),但Co3+为非磁的低自旋态(S=0)。因此,考虑材料特殊的电荷有序分布,PbCoO3可表述为A 位与B 位同时有序的四重钙钛矿,空间群为立方Pn3,如图12(a)所示。由于B 位S=0 的低自旋Co3+离子与高自旋Co2+离子的盐岩型有序分布,在磁性上如果只考虑具有S=3/2 的磁性Co2+,它们将构成四面体的几何阻挫。通常情况下,这种几何阻挫磁性体系不具有长程磁有序。然而,有趣的是,PbCoO3( Pb2+Pb43+Co22+Co32+O12)在较低温度下展现了2 个长程反铁磁相变,相变温度大约在8 和4 K,这种几何阻挫体系中的反常长程反铁磁有序行为可能与四面体轻微的结构扭曲相关,如图12(b)所示。

图12 PbCoO3 的(a)晶体结构、(b)磁化率和比定压热容(cp)[38]Fig.12 (a) Crystal structure, (b) magnetic susceptibility and constant pressure specific heat capacity (cp) of PbCoO3[38]
回顾PbCoO3苛刻的合成条件,不难想象 Pb2+Pb43+Co22+Co32+O12这种奇特的电荷组合形式对外界条件非常敏感。如果用压力进行调控的话,高压下预计可经历系列电荷相变与结构相变。基于上述考虑,Liu 等[39]对该物质开展了原位高压电阻、同步辐射X 射线衍射、发射光谱、吸收谱以及高压中子等系列测试,发现自旋态与电荷态敏感的压力存在依赖关系。随着压力逐渐增大至15 GPa,Co2+离子连续由高自旋态转变为低自旋态,如图13(a)所示。虽然原位高压往往可以扩展能带宽度、降低电阻,但自旋态的改变使得PbCoO3的电阻随压力增大反常增大。当自旋态转变结束后,低自旋Co2+离子与反常高价态Pb4+离子之间发生电荷转移,电荷转移的积累效应导致材料在20 GPa 附近发生一级晶体结构相变,使材料由原来的立方晶系转变为四方晶系。由于电荷转移破坏了B 位电荷有序分布,因此当结构相变发生时,材料也伴随着绝缘体-金属化相变。Pb4+-Co2+的电荷转移导致低自旋Co2+(S=1/2)全部转变为绝缘的低自旋Co3+态(S=0),进而在30 GPa 时引起第2 次一级晶体结构相变以及再进入的绝缘化行为(金属-绝缘体转变)。这也使得自旋态与电荷转移两种电子相变首次在单相材料中被同时发现,并且导致晶格、电荷、自旋、轨道等多个自由度的急剧改变,如图13(b)所示。
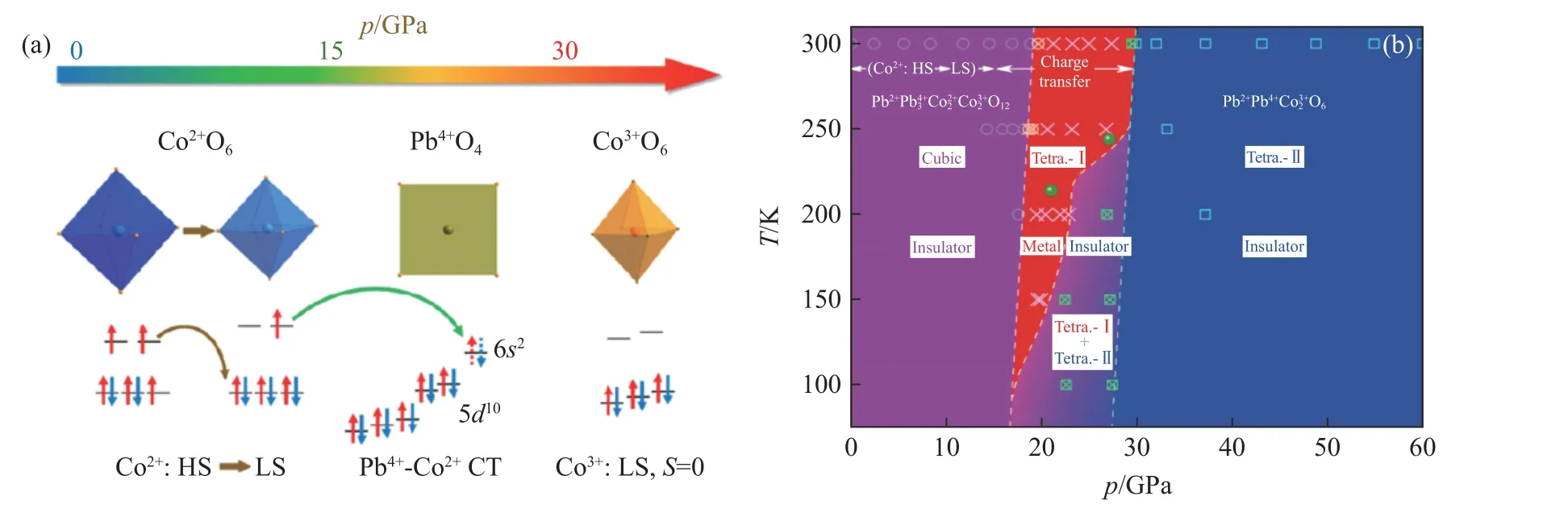
图13 (a) PbCoO3 中自旋和价态随压力的演化,(b) PbCoO3 的压力温度相图[38]Fig.13 (a) Schematic view of the origin of pressure induced spin state transition and charge transfer in PbCoO3;(b) pressure and temperature dependent phase diagram of PbCoO3[38]
6 PbNiO3 的高压制备和物性研究
与前面介绍的几种Pb 系钙钛矿材料不同,PbNiO3的实验制备和结构解析难度并不大。日本学习院大学的Inaguma 教授利用高温高压实验手段在3 GPa、1 073 K 条件下制备了具有Pnma结构的正交钙钛矿PbNiO3,发现在常压下退火能够使其很快转变为更低对称性的LiNbO3(R3c)结构,如图14 所示。X 射线光电子能谱(XPS)测试发现其电子态为Pb4+Ni2+O3。磁性测试表明,这2 种材料都表现为反铁磁性,转变温度分别为225 和205 K;电阻测试表明,2 种结构的PbNiO3均表现为半导体行为。从PbNiO3的研究可以发现,由于Pb4+相对Ni2+的离子半径较小,导致其容忍因子已接近钙钛矿成相的下限,因此,在常压条件下退火就能转变为对称性更低的铌酸锂结构[40]。
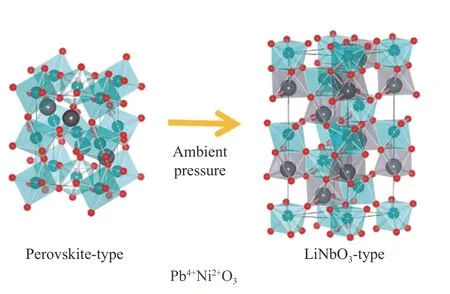
图14 PbNiO3 的晶体结构及其温度诱导的结构相变[40]Fig.14 Crystal structure and temperature induced structure phase transition of PbNiO3[40]
7 结 论
PbMO3(M=3d过渡族金属)随着M 元素原子序数、d轨道能级深度的逐渐增加,其晶体结构、电子态、磁性和电输运性质表现出一系列演化行为,同时这些行为在高压调制下还出现了结构相变、电荷转移和绝缘体-金属相变等诸多奇异的性质,如图15 所示。此外,由于3d元素关联特性较强,可以将其界定为一种较为典型的关联材料研究体系,用以验证与关联体系相关的计算方法。目前,该材料体系中还有2 种元素(Zn 和Cu)的钙钛矿或衍生结构没有通过实验合成出来。笔者曾经尝试合成含Zn 的铅系过渡族金属氧化物,但只在6 GPa 压力条件下得到铌酸锂结构的ZnPbO3[41],同时高压衍射实验发现在高压下ZnPbO3可以转化为正交钙钛矿结构,因此能否在更高压力下直接利用高温高压技术合成钙钛矿结构尚需进一步证实。尝试合成PbCuO3的探索过程中发现,在现有实验范围内只能得到一个阳离子反占位极为严重的烧绿石晶体结构,并且在15 GPa 左右条件下还没有出现新的结构。从PbCoO3的研究经验看,在较低压力下可以合成A/B 位反占位严重的烧绿石结构[42],在更高压力温度下才转变成钙钛矿结构,因此,在更高压力下合成PbCuO3钙钛矿结构还是极有希望的,这也是该体系研究中接下来需要努力的方向。
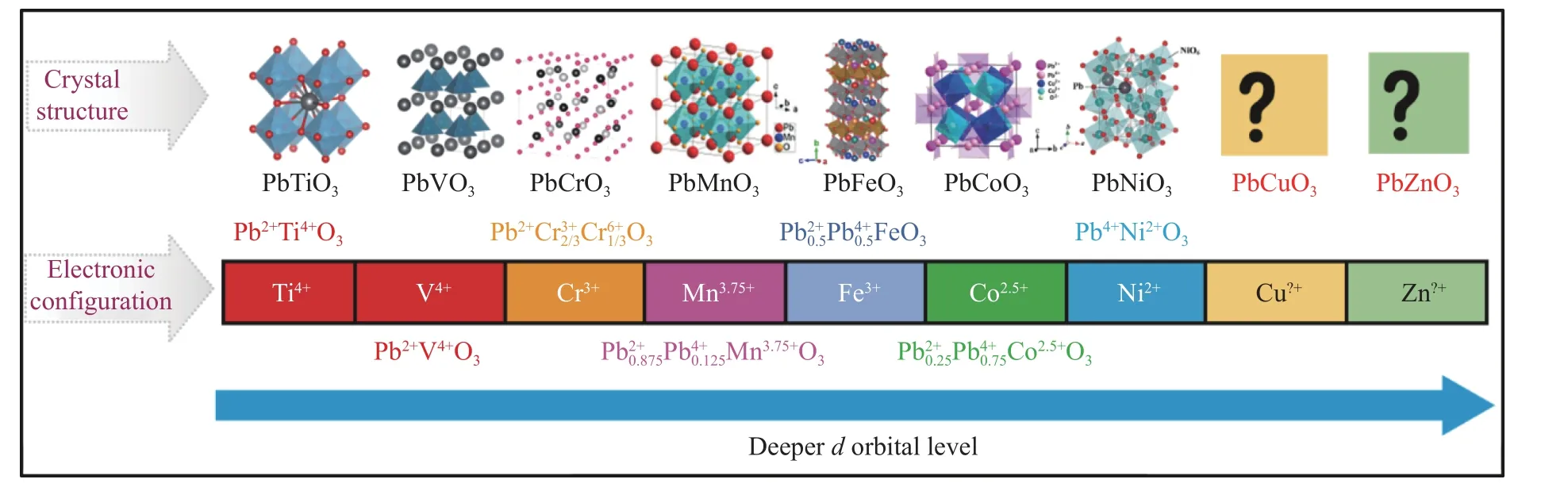
图15 PbMO3 系列钙钛矿材料的晶体结构和电子态的演化示意图Fig.15 Schematic diagram of crystal and electronic configuration of PbMO3 series compounds
感谢中国科学院物理研究所赵建发、刘哲宏、叶旭斌、龙有文和靳常青研究员以及北京理工大学李翔教授等在本文写作中给予的大力支持和帮助。
