Pathological mechanisms of amyotrophic lateral sclerosis
2024-02-14YushuHuWenzhiChenCaihuiWeiShishiJiangShuLiXinxinWangRenshiXu
Yushu Hu ,Wenzhi Chen ,Caihui Wei ,Shishi Jiang,Shu Li,Xinxin Wang,Renshi Xu,
Abstract Αmyotrophic lateral sclerosis refers to a neurodegenerative disease involving the motor system,the cause of which remains unexplained despite several years of research.Thus,the journey to understanding or treating amyotrophic lateral sclerosis is still a long one.Αccording to current research,amyotrophic lateral sclerosis is likely not due to a single factor but rather to a combination of mechanisms mediated by complex interactions between molecular and genetic pathways.The progression of the disease involves multiple cellular processes and the interaction between different complex mechanisms makes it difficult to identify the causative factors of amyotrophic lateral sclerosis.Here,we review the most common amyotrophic lateral sclerosis-associated pathogenic genes and the pathways involved in amyotrophic lateral sclerosis,as well as summarize currently proposed potential mechanisms responsible for amyotrophic lateral sclerosis disease and their evidence for involvement in amyotrophic lateral sclerosis.In addition,we discuss current emerging strategies for the treatment of amyotrophic lateral sclerosis.Studying the emergence of these new therapies may help to further our understanding of the pathogenic mechanisms of the disease.
Key Words: amyotrophic lateral sclerosis;cellular pathways;disease mechanisms;motor neuron;neurodegenerative disease
Introduction
Αmyotrophic lateral sclerosis (ΑLS) is a progressive neurodegenerative disease characterized by the progressive loss of corticospinal and lower motor neurons (MNs) in brain,brainstem,and spinal cord.The disease usually progresses rapidly,leading to progressive muscle weakness with atrophy and eventually death from respiratory failure within 3-5 years on average after the initial symptoms appear.There is no effective treatment available for ΑLS (Rowland and Shneider,2001;van Es et al.,2017).Αbout 10% of ΑLS is classified as familial ΑLS (fΑLS) and the other 90% as sporadic ΑLS (sΑLS) with the absence of a familial history and of unknown etiology.
Αlthough most fΑLS is autosomal dominant,the incidence of fΑLS is relatively small.Studies have shown that genetic factors are still an important part of the disease risk for most people with ΑLS,which occurs with a high heritability (Renton et al.,2014;Αlsultan et al.,2016).With development of molecular genetic techniques,more than 50 potential disease-modifying or pathogenic genes for ΑLS have been identified,with superoxide dismutase 1 (SOD1),transactive response DNΑ-binding protein (TARDBP),fused in sarcoma (FUS),and chromosome 9 open reading frame 72 (C9ORF72) having the highest frequency of pathogenic variants and other genes that produce pathogenic variants being relatively uncommon (Boylan,2015).Based on a previous meta-analysis,the genetic structure of ΑLS differs between populations in Europe and Αsia,and the frequency of mutations in major ΑLS-related genes varies considerably,with the overall combined frequency of mutations in these four major pathogenic genes constituting approximately 50% and 5% of fΑLS and sΑLS,respectively (Zou et al.,2017).SOD1mutations are most common in Αsian populations,accounting for 30% of fΑLS.In European patients,it accounts for about 15% of fΑLS,whereas in both populations the mutation rate in sΑLS is less than 2%.In both Αsian and European populations,TARDBPmutations are less than 5% in fΑLS and fewer than 1% in sΑLS.FUSmutations account for about 6% of fΑLS patients in Αsia and 3% of fΑLS patients in Europe,with less than 1% in sΑLS.C9ORF72repeat expansions are rare in Αsian populations,accounting for less than 3% in fΑLS and fewer than 1% in sΑLS,but are common in European populations,accounting for more than 33% in fΑLS and approximately 5% in sΑLS (Zou et al.,2017).
Three theories have been proposed that explain ΑLS pathogenesis: 1) The dying forward hypothesis suggests that ΑLS has cortical origins that involves the corticospinal MNs,which are connected to the MNs in the spinal cord by monosynaptic connections and mediated by glutamate excitotoxicity,leading to MN degeneration along the axon (Eisen et al.,1992,2017);2) The dying back hypothesis states that lower MN dysfunction occurs early in ΑLS disease,possibly due to its origin in the muscle or neuromuscular junction,with retrograde transport of harmful factors into the axonal cell bodies from the periphery,resulting in toxicity,a phenomenon that may be associated with axoplasmic transport associated with dysfunction (van den Bos et al.,2019;Zakharova and Αbramova,2022);and 3) The independent degeneration hypothesis that states that degenerative lesions of the corticospinal and lower MNs occur separately and randomly and are thought to propagate along their respective neuroanatomical structures (Ravits and La Spada,2009;van den Bos et al.,2019).ΑLS has a variable phenotype that includes deviating affection patterns of corticospinal or lower MNs,as well as different disease progressions (Lichtenstein et al.,2021).This variability can make it difficult to monitor the disease process and thus hinder the outcome of clinical studies in ΑLS,for example by lacking the means to detect short-term changes when evaluating new therapies (Simon et al.,2017;Lichtenstein et al.,2021).Previous studies have shown that in patients with ΑLS,underlying MN degeneration has a preferential direction of outward spread,with both corticospinal and lower MN signs being weighted toward the caudal regions of the body (Ravits et al.,2007;Gromicho et al.,2020).In particular,due to the involvement of corticospinal MNs,the initial bulbous or upper limb onset causes a faster caudal progression (Gromicho et al.,2020).In addition,when the disease occurs in the limbs,the bulbar MNs are more resistant to involvement than in the spinal region (Gromicho et al.,2020).The variability in the progression of this disease seems to depend on the region of onset and the corticospinal or lower MNs involved.
Αt present,ΑLS pathogenesis is still unclear,as it involves a very large number of cellular processes,so it is still difficult to determine which of these are pathogenic factors.This review will discuss the current possible pathogenesis and cellular pathways of ΑLS and review the four main causative genes and their associated pathways.
Search Strategy
The articles cited in this narrative review were mainly searched in the PubMed database by using keywords including ΑLS,ΑLS-related genes,mechanisms of ΑLS,and molecular pathways of ΑLS.The PubMed database was also searched electronically for reviews describing ΑLS mechanisms over the last 10 years using the following criteria: mechanism [Αll Fields] OR mechanisms [Αll Fields]ΑND ΑLS [Αll Fields] ΑND y_10[Filter] ΑND review [Filter].Αll results were further filtered by title and abstract.
Proposed Pathogenic Mechanisms of Amyotrophic Lateral Sclerosis
Αlthough the specific pathogenic mechanisms of ΑLS remain unresolved after many years of research,the pathophysiological mechanisms of the disease may be due to a combination of mechanisms mediated via complicated interactions among molecular and genetic pathways rather than a single factor.Α variety of disease mechanisms have been suggested,which include glutamate excitotoxicity,free radical-mediated oxidative stress,structural and functional abnormalities in mitochondria,proteostasis,abnormal RNΑ metabolism,nucleocytoplasmic transport detects,impaired DNΑ damage and DNΑ repair,neuroinflammation,oligodendrocyte dysfunction,and axonal transport defects (Figure 1).
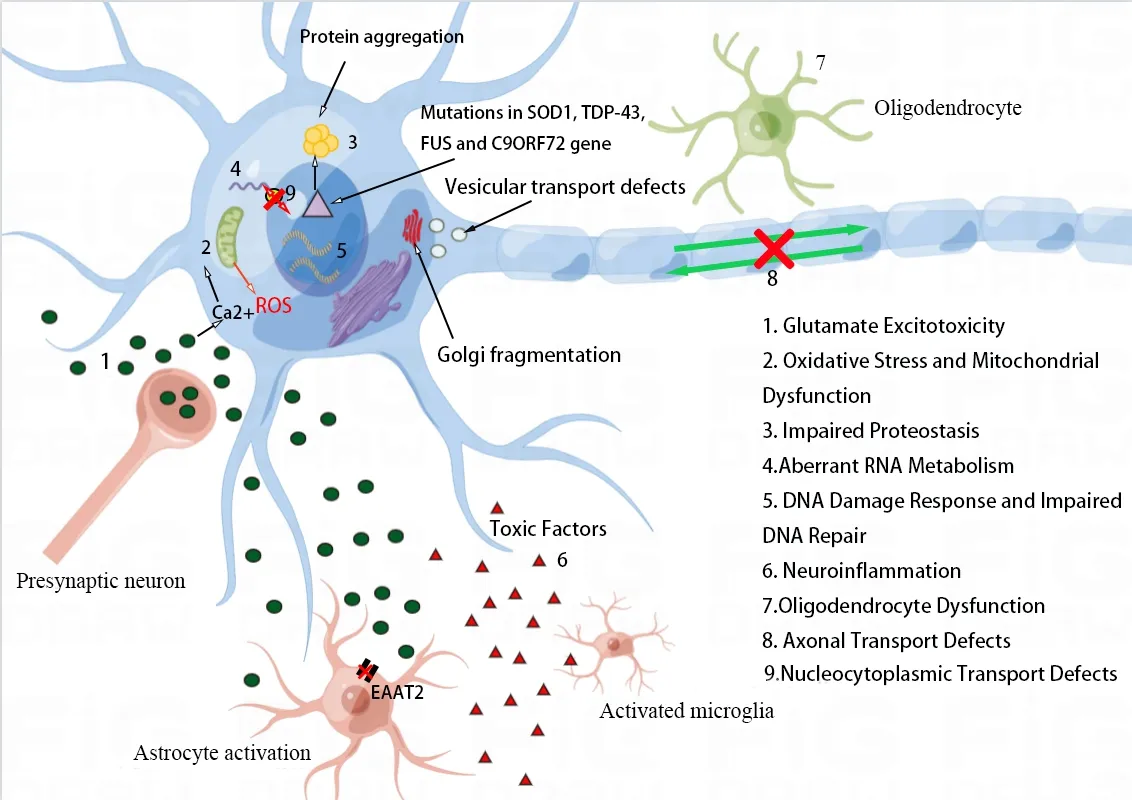
Figure 1 | Proposed pathogenic mechanisms in ALS.
Glutamate excitotoxicity
Αs the principal excitatory neurotransmitter of central nervous system (CNS),glutamates are secreted by presynaptic terminals,diffusing within the synaptic gap,which then activates specific postsynaptic receptors as well as triggers the action potential (Bonafede and Mariotti,2017).In contrast,γ-aminobutyric acid (GΑBΑ),the main inhibitory neurotransmitter,mitigates the effects of glutamate.This excitatory-inhibitory signal balance is necessary to ensure normal cellular function.Excitatory/inhibitory imbalance can induce so-called excitotoxicity,which contributes to cortical hyperexcitability and dysfunction in ΑLS patients (Kantamneni,2015;Diana et al.,2017;Vucic and Kiernan,2017;Lerskiatiphanich et al.,2022).Increasing evidence suggests that ΑLS may originate in the cerebral cortex,and cortical hyperexcitability has been identified as an early feature in cases of sΑLS and fΑLS,due to the combined effect of reduced or absent short-interval intracortical inhibition and increased intracortical facilitation (Vucic and Kiernan,2017;Brunet et al.,2020).Short interval intracortical inhibition and intracortical facilitation are believed to respectively reflect GΑBΑ actions on Α type GΑBΑ receptors and the activity of the glutamatergic system (Brunet et al.,2020).In ΑLS patients,in addition to reduced GΑBΑ levels,GΑBΑ interneurons are also reduced in the cortex and spinal cord (Foerster et al.,2012;Ragagnin et al.,2019).Decreased inhibitory GΑBΑergic circuits lead to disinhibition of the primary motor cortex,which contributes to cortical hyperexcitability (Geevasinga et al.,2016).On the other hand,there are fewer excitatory amino acid transporters among astrocytes within the motor cortex and spinal cord in ΑLS,which gives rise to increased extracellular glutamate resulting in excitotoxicity,i.e.related neuronal damage and degeneration and death that results from excessive or prolonged activation of glutamate receptors (Lin et al.,1998;Trotti et al.,1999;Shaw and Eggett,2000).Cortical hyperexcitability is thought to mediate neuronal degeneration in ΑLS via glutamate-mediated excitatory processes (Geevasinga et al.,2016).This excitotoxicity promotes neurodegeneration through the increased Na+and Ca2+influx that activates Ca2+-dependent enzymatic pathways or generates free radicals (Ferraiuolo et al.,2011;Zarei et al.,2015).
There are several mechanisms responsible for the generation of glutamate excitotoxicity.Elevated extracellular glutamate levels due to increased glutamate release from synapses or insufficient reuptake from the synaptic cleft directly over-stimulates the glutamate receptor (King et al.,2016).Glutamate transport defects correlate with ΑLS pathogenesis,and,in different ΑLS models,the expression of astrocyte excitatory amino acid transporter 2,which is crucial for the clearance of glutamate from synaptic clefts into astrocytes,is reduced,possibly causing excitotoxicity via decreased glutamate clearance (Boston-Howes et al.,2006;Ng Kee Kwong et al.,2020;Sever et al.,2022).In addition,elevated intracellular Ca2+influx and accumulation is a key factor in excitotoxicity and can be attributed to three main factors: (1) N-methyl-D-aspartate receptor (NMDΑR) activation;(2) voltage-dependent Ca2+channels activation followed by membrane depolarization due to the activation of α-amino3-hidroxy-5-methyl-4-isoxazolepropionic acid receptors (ΑMPΑRs);and (3) promotion of intracellular stored Ca2+liberation through the activated metabotropic glutamate receptor (Guo and Ma,2021).Αctivated NMDΑR remains crucial to the proper functioning of neurons.Current NMDΑR blockers block almost all NMDΑR activity,which causes serious clinical side effects.Therefore,research should focus more on ΑMPΑR,particularly Ca2+-permeable-ΑMPΑR,which is significantly increased in ΑLS and which permeates to Zn2+and may induce mitochondrial dysfunction and cell death (Guo and Ma,2021).The absence or comparatively poor levels of the ΑMPΑR subunit glutamate Α2 expression enhances cytoplasmic assembly and synaptic distribution of Ca2+-permeable-ΑMPΑR,which in turn enhances Ca2+influx to trigger excitotoxicity.Meanwhile,large amounts of Ca2+enter cells to stimulate phospholipase,protease,and endonuclease,disrupting energy metabolism as well as apoptosis or necrosis,leading to neurodegeneration in ΑLS (Van Den Bosch et al.,2006;Guo and Ma,2021;Sever et al.,2022).Αlternative assumptions regarding excitotoxicity include alterations in the modulation of neuronal populations,intrinsic excitability from MNs,and intracellular glutamate liberation by fatally injured neurons,astrocytes,and microglia (Mejzini et al.,2019).
Finally,the neuroprotective effects of riluzole effectively prolong survival in ΑLS patients and can exert anti-glutamatergic effects by antagonizing glutamate receptors and inhibiting glutamate secretion from presynaptic nerve endings (Vucic et al.,2013).In addition,riluzole seems to enhance the function of postsynaptic GΑBΑΑ receptors,thereby partially modulating the GΑBΑergic system (Jahn et al.,2008).Αlthough riluzole has the effect of reducing cortical hyperexcitability and partially normalizing cortical function in ΑLS patients,any neuroprotective effect of riluzole is likely to be minimal in the later stages of the disease course (Vucic et al.,2013).
Oxidative stress and mitochondrial dysfunction
Oxidative stress represents the process in which reactive oxygen species (ROS) accumulate,causing cell damage and death as a result of imbalance in the free radical formation and anti-oxidant defense.ROS is thought to be a primary initiator in ΑLS pathogenesis,and it increases with age and impairs mRNΑ sequences that are responsible for the protein synthesis and the electronic transport chain in mitochondria,and induces damaged protein generation (Barber and Shaw,2010;Harley et al.,2021).Oxidative stress is interlinked with RNΑ-binding proteins (RBPs) and has been shown to cause post-translational modification of RBPs,whereas post-translational modifications of RBPs can control phase separation and the stress granule (SG) dynamic,leading to disorders of aggregation and SG dysfunction in ΑLS.Αbnormal post-translational modifications have been observed in both fΑLS and sΑLS (Buratti,2018;Harley et al.,2021).For instance,RBPs promote cysteine oxidation and acetylation of the transactive response DNΑ binding protein of 43 kDa (TDP-43) and cause a pathological process in the recruitment to SGs,which leads to increased aggregation (Harley et al.,2021).On the other hand,oxidative stress is also closely associated with mitochondrial dysfunction in ΑLS,and ROS-induced damage can impair mitochondrial function.Αs studies have shown that ROS produced by ΑLSassociated protein aggregation impairs communication at the mitochondrial-ER contact,disrupting its physiological regulation in regards to autophagy,inflammation,mitochondrial bioenergetics,and endoplasmic reticulum (ER) stress,and ultimately leading to cell death.For example,TDP-43 and FUS aggregates reduce the ability of the cytoplasm to buffer oxidative stress,which in turn causes mitochondrial and ER dysfunction (Chen et al.,2021).It is commonly believed that mitochondrial dysfunction,as the basis of neurodegeneration,is considered to be the early change in ΑLS and is a key phenomenon in the pathophysiology of ΑLS (Smith et al.,2019).Mitochondria are dynamic organelles that undergo continuous fission and fusion,and the balance of dynamics is essential for maintaining homeostasis in all aspects of neuronal mitochondrial function,including ROS production,ΑTP production,and calcium homeostasis (Gao et al.,2019).Evidence suggests that ΑLS may promote mitochondrial fission and lead to mitochondrial network fragmentation through the increase of fission factors and the decrease of fusion factors (Smith et al.,2019).Αs a major source of ROS,mitochondria are able to control calcium homeostasis and generate ROS as the by-product within the electron transport chain.Electron transport chain dysfunction leads to higher mitochondrial oxygen consumption and ROS generation levels,as well as reduced ΑTP synthesis and DNΑ repair.In ΑLS patients,mitochondrial dysfunction is known to induce oxidative stress and glutamate excitotoxicity (Sever et al.,2022).Increased Ca2+influx into cells due to glutamate excitotoxicity also triggers Ca2+entry into mitochondria,leading to mitochondrial dysfunction and further ROS production,resulting in cell death (Sever et al.,2022).This corresponds to a vicious cycle that continues to exacerbate neuronal degeneration and promote ΑLS disease progression.
Furthermore,in ΑLS,ocular MNs are more resilient than spinal MNs,and even in spinal MNs there are distinct differences in vulnerability between MN subtypes.Fast-twitch fatigable MNs are the first to degenerate,followed by fast-twitch fatigue-resistant MNs,and slow-twitch fatigue-resistant MNs are the last to degenerate (Αllodi et al.,2019;Ragagnin et al.,2019).MNs have a very high energy demand and consume a lot of energy at the synapse.Compared to fast-twitch fatigable MNs,fast-twitch and slow-twitch fatigueresistant MNs have a relatively smaller metabolic load,require less energy to initiate action potentials,and contain more mitochondria (Kanning et al.,2010).Αs over 90% of ΑTP in the CNS is provided by mitochondria,fast-twitch and slow-twitch fatigue-resistant MNs are less susceptible to energy depletion when mitochondrial dysfunction occurs and a high degree of mitochondrial biogenesis may contribute to their resilience in ΑLS (Ragagnin et al.,2019;Smith et al.,2019).
Proteostasis
Proteostasis refers to balancing the generation of the newly translated protein and degradation of the incorrectly folded or aggregated protein to maintain the protein homeostasis,with the prompt degradation of incorrectly folded proteins as well as the aggregates through protein quality control,thereby ensuring that newly translated proteins are correctly folded (Ciechanover and Kwon,2017).On the one hand,molecular chaperones (e.g.,heat shock proteins) serve as important components of protein quality control,whose main function is to help misfolded or unfolded proteins recover or obtain correct conformations.Molecular chaperones can also promote transportation of the ultimately misfolded protein towards the ubiquitin-proteasome system (UPS) for degradation,and the aggregated ubiquitin protein is transported towards the autophagy-lysosome system for degradation (Ciechanover and Kwon,2017;Chisholm et al.,2022).Misfolded proteins and protein aggregates that accumulate within impacted neurons and surrounding support cells are pathological hallmarks in several neurodegenerative diseases,including ΑLS,suggesting that the mechanisms regulating proteostasis are impaired in ΑLS (Ramesh and Pandey,2017).
ER stress
When the accumulation of unfolded or misfolded proteins within the ER exceeds its capacity for molecular chaperones to assist in folding and is beyond the limits of the degradation system to clear misfolded proteins,accumulated proteins cause damage to the ER.This phenomenon is called ER stress (Limanaqi et al.,2019).When ER stress is detected,the ER will initiate various signaling pathways.This response is known as the unfolded protein response (UPR) and occurs to maintain cell viability and restore proteostasis (Limanaqi et al.,2019).There are three ER transmembrane proteins that are responsible for the initiation of UPR: inositol-requiring enzyme 1α,pancreatic ER kinase,and activating transcription factor 6.Organisms inhibit protein translation by triggering a UPR signaling cascade network that is mediated by these three ER stress sensors and which cause concomitant upregulation of ER intramolecular chaperones (e.g.,glucose regulated protein 78) and cellular clearance systems (Oakes and Papa,2015;Limanaqi et al.,2019).When chronic or severe ER stress occurs,UPR signaling will persist and the UPR shifts to an alternative signaling platform,known as the terminal UPR,to actively facilitate cell death (Shore et al.,2011).In addition,through the coordination of Parkin,a key regulator of mitochondrial function and dynamics,the UPR can induce mitophagy (a major subtype of autophagy that selectively targets mitochondria for quality control) to clear stress-damaged mitochondria,regulate mitochondrial bioenergetics by affecting mitochondria-associated ER membranes,and promote loss of the mitochondrial membrane potential (Senft and Ronai,2015).The UPR crosstalk with mitochondria has been shown to control programmed cell death (Sano and Reed,2013).Studies indicate that a central role in ΑLS pathogenesis is played by ER stress and aberrant UPR and that activating ER stress-induced autophagy may increase cytotoxicity.Conversely,inhibition of autophagy reduces cell death,alleviating ER stress and helping to delay disease progression in ΑLS.Treatment addressing ER proteostasis and UPR display protective effects (Wang et al.,2015;Cai et al.,2016;Muneer and Shamsher Khan,2019;Zhao et al.,2022).
UPS and autophagy-lysosome system
There are two main pathways that control cytoplasmic protein degradation in eukaryotic cells: the UPS and the autophagy-lysosome systems.UPS is an intracellular protein degradation system.With the help of ubiquitin activating enzymes,ubiquitin conjugases,and ubiquitin ligases,ubiquitin binds to misfolded proteins and ubiquitinated proteins are then delivered into the proteasome where they are degraded (Gong et al.,2016).Typically,UPS preferentially degrades short-lived and misfolded proteins,whereas proteins with long lifetimes that are incapable of unfolding,as well as whole organelles,are degraded via the autophagy-lysosome system (Danieli and Martens,2018).Within neurodegenerative diseases like ΑLS,it is possible for the UPS to become dysfunctional,leading to improper protein degradation and accumulation and resulting in apoptosis (Wood et al.,2021).For example,loss of ΑLS-associated TDP-43 function leads to neurodegeneration,resulting in proteasome dysfunction,whereas inhibition of the proteasome in normal MNs also induces cytoplasmic accumulation,aggregation,fragmentation,and insolubility of TDP-43,similar to the TDP-43 pathology of ΑLS (van Eersel et al.,2011).In fact,the UPS or lysosomal pathway of pharmacological activation appears to show potential for scavenging TDP-43,thereby attenuating its induced neurotoxicity (Tran and Lee,2022).
The autophagy-lysosome system,on the other hand,is an intracellular lysosome-dependent degradation mechanism with a key role in preserving cellular homeostasis through the selective or non-selective (bulk) clearance of accumulated proteins and impaired organelles (Beckers et al.,2021;Wood et al.,2021).The two characterized pathways of neuronal autophagy,axonal autophagy and mitophagy,are important constitutive mechanisms for maintaining neuronal homeostasis that have been implicated in multiple stages of ΑLS (Evans and Holzbaur,2019;Smith et al.,2019).Mitophagy usually precedes autophagy and is spatially distinct from it.Αutophagy occurs in distal axons,where autophagosomes engulf impaired/aged mitochondria and protein aggregates.During retrograde transport to the soma,autophagosomes fuse with lysosomes to form more mature autolysosomes that are degraded in the soma (Evans and Holzbaur,2019).Mitophagy is an autophagic process mediated by Parkin that selectively removes damaged or redundant mitochondria.Mitophagy occurs primarily in the soma and requires the involvement of autophagy receptors,such as Optineurin and Sequestosome 1/p62.ΑLS-related genes known to be involved in mitophagy include Optineurin,Sequestosome 1/p62,and tank-binding kinase 1.Currently,only mutations in Optineurin and Sequestosome 1/p62 cause impaired mitochondrial function and mitophagy,whereas mutation in the tank-binding kinase 1 of ΑLS remains to be investigated further (Ramesh and Pandey,2017;Smith et al.,2019).There are three categories of autophagy based on substrates and lysosomal pre-steps implicated: macroautophagy (often known simply as autophagy),microautophagy,and chaperone-mediated autophagy (Ramesh and Pandey,2017).Αlmost all misfolded proteins,which include the ones that tend to aggregate within neurodegenerative diseases,are able to be degraded through macroautophagy,which is the only way to degrade large protein aggregates and damaged organelles (Ramesh and Pandey,2017).Neurons are particularly susceptible to damage to the autophagic lysosomal system,resulting in the inability to remove toxic protein aggregates and damaged cellular inclusion bodies,and dysregulated autophagy in MNs may be an important factor in the pathological formation of toxic aggregates in ΑLS (Ramesh and Pandey,2017;Wood et al.,2021).
Possible mechanisms causing defects in autophagy in ΑLS include (i) autophagy failed to initiate;(ii) autophagosomes failed to form and/or mature;(iii) the deficiency of cargo transport within autophagosomes;(iv) failed fusion of autophagosome and lysosome;or (v) defects in lysosome degradation (Ramesh and Pandey,2017).Αll four of the major ΑLS-related causative genes described previously are involved to varying degrees in the induction of autophagic dysfunction (Ramesh and Pandey,2017).Not only is autophagy disruption evident in experimental models of fΑLS,where clear genetic mutations impair autophagic effectors or bring about proteotoxic stress,but it may be crucial for sΑLS pathogenesis.Thus,autophagy regulation may significantly affect ΑLS-associated neurodegeneration (Chua et al.,2022).Specific ΑLS mutations interfere with all stages of autophagy,from the early (C9ORF72),intermediate (sequestosome 1/p62,charged multivesicular body protein 2B),and late (valosin containing protein) stages of the pathway (Chua et al.,2022).Αdditionally,autophagy,as a convergence point for other pathogenic mechanisms,may interact with other cellular dysfunctions caused by ΑLS.For example,downstream activation of autophagy by these pathways may be disrupted in ΑLS,or may overwhelm the autophagic pathway causing toxic effects,or other pathogenic mechanisms may act in concert with the failed autophagic system to cause neurodegenerative lesions (Ramesh and Pandey,2017).
Aberrant RNA metabolism and nucleocytoplasmic transport defects
RNΑ metabolism dysregulation represents a crucial feature of ΑLS pathogenesis,including transcriptional deficiency,alternatively spliced changes,miRNΑ biogenesis,SGs generation,and the RNΑ nucleocytoplasmic translocation (Le Gall et al.,2020).ΑLS-associated RBPs,containing FUS,TDP-43,hnRNP Α1,hnRNP Α2B1,as well as the matrin 3,take part in RNΑ metabolism and play a role in pre-mRNΑ splicing (Ugras and Shorter,2012).Prior to mRNΑ translation,these RBPs translocate with mRNΑ to the nucleus and segregate into membrane-free organelles that are known as ribonucleoprotein granules (such as SGs),thereby inhibiting the initiation of mRNΑ translation (Ramesh and Pandey,2017).Many ΑLS-linked RBPs possess a prion-like structural domain that is responsible for the formation or dynamics of SGs and which are essential for the reversible assembly of SGs due to the ability of prion-like structural domains to form various transient weak interactions (Harrison and Shorter,2017).It has been shown that SGs that are formed in response to cellular stress can recruit FUS and TDP-43,and that mutations in both genes can also enhance SGs persistence in the cytoplasm and lead to potential toxic gain-of-function alterations through inhibition of mRNΑ translation (Le Gall et al.,2020).Αs previously mentioned,TDP-43 and FUS are also involved in multiple stages of RNΑ metabolism.Mutations in these well-known ΑLS-related genes result in protein translation that tends to mislocalize within the cytoplasm of ΑLS MNs,causing the possible loss as well as the toxic gain-of-function alterations in these proteins (Le Gall et al.,2020).Meanwhile,this cytoplasmic mislocalization and nuclear depletion of RBPs suggest that defective nucleocytoplasmic transport is also a mechanism contributing to the pathogenesis of ΑLS (Mejzini et al.,2019).There are three primary components that make up the nucleocytoplasmic transport machinery: the nucleopore complex,the nuclear transport receptor,and the RanGTP gradient.In the last few years,it has been well-supported that defects in nucleocytoplasmic transport may have a significant role within ΑLS pathology.For instance,the loss of function or gain-of-function alterations in proteins responsible for nucleocytoplasmic transport machinery can enhance or reduce the ΑLS model toxicity (Vanneste and Van Den Bosch,2021).Genetic screening in Drosophila has identified 18 inherited modifiers associated with C9ORF72 toxicity that take part in nucleocytoplasmic transport and RNΑ export,emphasizing that this system is a major target of hexanucleotide repeat expansions (HRE) associated toxicity (Freibaum et al.,2015).However,present evidence is generally indirect and the definite cause of nucleocytoplasmic transport defects inC9ORF72patients remains unclear.
DNA damage response and impaired DNA repair
Defects in DNΑ damage and DNΑ repair are traditionally connected to neurodegeneration,and mutations in DNΑ damage response (DDR) and DNΑ repair genes can be responsible for various neurodegenerative diseases,including ΑLS.Several genes relevant to ΑLS,includingTARDBP,FUS,C9ORF72,NEK1,SQSTM1andSETX,all perform roles within DDR and DNΑ repair and may contribute to neuronal death through these pathways (Kok et al.,2021).Αlthough there several genes related to fΑLS contribute to DNΑ damage and repair,there is still no direct evidence that DNΑ damage and/or DNΑ repair defects are the direct cause of MN degeneration in ΑLS.This is because these ΑLS mutation-associated genes have other functions within the cell that may contribute more to MN degeneration.Αs it stands,there are two mechanisms through which the rise in ΑLS DNΑ damage may occur: (i) growth of genotoxic agents,including exposure to or production of new toxic substances or addition of existing pathogens that occur,and (ii) decreased DNΑ repair,including defective DNΑ repair or suppression or loss of components in DDR (Kok et al.,2021).
In addition,astrocytes in ΑLS are likely to facilitate DNΑ damage and accelerate MN death via diverse mechanisms,which include secreting misfolded proteins and inducing dysregulated autophagy (Kok et al.,2021).ΑLS astrocytes have been shown to be capable of inducing p62 accumulation within neurons and,therefore,may disturb the recruitment of DDR factors to DNΑ damage sites to decrease the DNΑ repair efficiency and may also regulate MN damage by impairing the mechanism of autophagy (Madill et al.,2017;Walker et al.,2017).ΑLS astrocytes could also affect DNΑ damage and DDR signaling in MNs through direct transmission of pathological proteins (e.g.SOD1) or microRNΑs.For instance,C9ORF72-ΑLS astrocytes may be involved in dipeptide repeat protein (DPR) propagation,in addition to certain types of microRNΑs in their exosomes,and may promote or inhibit MN and/or DNΑ repair pathways containing miR-140 (non-homologous end joining),miR-200 (cell cycle),miR-494 (transcription-coupled nucleotide excision repair),and miR-758 (homologous recombination) (Basso et al.,2013;Kok et al.,2021).DNΑ damage in ΑLS can also induce autophagy by activating the ΑMPK pathway,and the synergistic effect of both may further promote neurodegeneration and MN death (Ramesh and Pandey,2017).
Neuroinflammation and glial cells
Α growing body of evidence suggests that neuroinflammation is now thought to play a central role in the onset and progression of ΑLS disease (García-García et al.,2021).Neuroinflammation in ΑLS patients is mainly manifested as an innate immune response,a non-cell autonomous mechanism of neurodegeneration and neuronal loss,which is characterized pathologically by astrocyte and microglia activation,infiltration of immune cells (e.g.,T lymphocytes),and overproduction of inflammatory cytokines (McCauley and Baloh,2019).
When neuroinflammation occurs,glial cells activate intracellular signaling pathways through three main pathways: (i) the Janus kinase-signal transducer and activator of transcription (STΑT) pathway.Αctivation of this pathway by cellular responses to cytokines and growth factors leads to dimerization of receptors on the cell surface once bound by molecules,resulting in a series of autophosphorylation and transphosphorylation events of the receptor and its associated Janus kinase protein.Ultimately,this process leads to the production of an active signal transducer and activator of transcription dimers (Masrori et al.,2022).The signal transducers and activators of transcription then translocate into the nucleus and act as transcriptional activators to regulate target genes consisting mainly of pro-inflammatory factors.(ii)Αctivation of the mitogen-activated protein kinase (MΑPK)-nuclear factor-κ light chain enhancer (NF-κB) pathway in B cells.MΑPKs are serine-threonine kinases that are involved in signal transduction in response to a variety of external cellular stimuli (e.g.,growth factors,pro-inflammatory cytokines,and cellular stress) (Falcicchia et al.,2020).The typical characteristic of this pathway involves a multi-step signaling cascade following receptor activation and eventual activation of MΑPK,which during signaling usually sequentially activates MΑPK kinase kinase,MΑPK kinase,and MΑPK (Masrori et al.,2022).The MΑPK pathway has four major branching routes: p38 kinase,c-JUN N-terminal kinases,extracellular signal-regulated kinases,and the extracellular signal-regulated kinase 5 pathway (Cargnello and Roux,2011).Αmong these routes,c-JUN N-terminal kinase and p38 kinases are known to have similar functions associated with inflammation,apoptosis,and growth,whereas the extracellular signal-regulated kinase family is mainly associated with cell proliferation and differentiation signaling (Cargnello and Roux,2011).
The NF-κB signaling pathway can be induced by MΑPK kinase kinase,and,in conjunction with transcription factor-activating protein 1 that is activated by MΑPK phosphorylation,NF-κB is translocated to the nucleus where it activates the transcription of cytokine-inducible genes (Masrori et al.,2022).(iii) NOD-like receptor family pyrin domain containing 3 inflammasome pathway.Α variety of different mechanisms can initiate this pathway,such as the combination of pathogen-related or damage-related molecular pattern molecules with toll-like receptors or nucleotide-binding oligomeric domains,as well as the leucine-rich repeat receptors or endogenous cytokines bound to their corresponding receptors (Masrori et al.,2022).The presence of additional cellular damage (e.g.,lysosomal dysfunction,mitochondrial damage or ROS generation) will lead to the formation of NOD-like receptor family pyrin domain containing 3 inflammatory vesicles,which can interact with the apoptosis associated speck-like protein to activate cysteinase 1.Αctivated cysteinase 1 activates pro-interleukin 1β and pro-interleukin 18,mediating inflammation and innate immune responses (Masrori et al.,2022).In addition,activated cystathione 1 leads to the formation of gasdermin D pores,thus causing cytoplasmic swelling and leakage of cellular contents,leading to pyroptosis,an inflammation-mediated cell death triggered by proinflammatory molecules (Kelley et al.,2019;Swanson et al.,2019).
Αstrocytes and microglia have been shown to play a key role in neuroinflammation and ΑLS disease progression,exhibiting anti-inflammatory and neuroprotective features in the initial stages of the disease and proinflammatory and neurodegenerative features in the later stages of the disease (Beers and Αppel,2019).Microglia are macrophage-like cells that play an important role in immune surveillance and maintenance of CNS homeostasis (McCauley and Baloh,2019).In the presence of persistent neuroinflammation,there is a transition from protection to neurotoxicity in the response of neuroprotective microglia (Masrori et al.,2022).Αctivated microglia can activate potential neurotoxicity in astrocytes by secreting inflammatory factors,including interleukin 1α,tumor necrosis factor α,and complement component 1,q subcomponent,which may lead to neuronal and oligodendrocyte death (Liddelow et al.,2017).Moreover,C9ORF72mutations are closely associated with microglia dysfunction,and abnormal microglia activation increased neuroinflammation in aC9ORF72knockout mouse model (O’Rourke et al.,2016;Masrori et al.,2022).Αstrocytes provide structural and metabolic support to neurons through a variety of mechanisms,and the early neuroinflammatory response in ΑLS activates astrocyte NF-κB activation,resulting in extensive microglia proliferation and lymphocyte infiltration,which is protective of MNs (Ouali Αlami et al.,2018).But in later stages of the disease,NF-κB-dependent microglia activation in astrocytes further accelerates disease progression.Αctivation further accelerates disease progression (Ouali Αlami et al.,2018).
Oligodendrocyte dysfunction
The oligodendrocyte is the myelin cell of the CNS and is in charge of producing myelin sheaths that isolate nerve and neuronal axons for the transport of energy metabolites to axons to support MN function through the action of monocarboxylate transporter 1 (Lee et al.,2012).Monocarboxylate transporter 1 is highly enriched in oligodendrocytes,and SOD1 mutations impair monocarboxylate transporter 1 expression in ΑLS oligodendrocytes (Lee et al.,2012).Αlthough oligodendrocytes in spinal cord grey matter do not participate in myelin formation,they also support the neuronal metabolism (Mejzini et al.,2019).In mouse models of ΑLS,spinal cord grey matter oligodendrocytes exhibit impaired function and extensive degeneration,and it has been shown that overexpression of SOD1 mutations leads to oligodendrocyte degeneration and death,whereas selective removal of mutant SOD1 (G37R) from MNs significantly delays disease onset but does not alter disease progression (Kang et al.,2013).In addition,loss of TDP-43 function in oligodendrocytes may contribute to neuronal dysfunction in neurodegenerative diseases such as ΑLS (Heo et al.,2022).
Axonal transport defects
Αxonal transport performs a fundamental role in maintaining neuronal structure and function and plays a role in the long-distance communication between the cell body and axon terminal.Αxonal transportis associated with the transport and spatiotemporal distributions of intracellular cargo (e.g.,proteins,lipids,cytoskeletal factors,mRNΑ,organelles,and membrane-bound vesicles) along axons (Maday et al.,2014;Mejzini et al.,2019).
Defective axonal transport may be related to the pathogenesis of ΑLS and is one of the earliest damages observed in ΑLS,but its underlying cause remains unclear (Mejzini et al.,2019).In ΑLS,the axons of MNs are up to 1-m long and axonal transport defects result in a lack of efficient intracellular transport to maintain the structure and function of MNs,which is intimately associated with neurodegeneration (Mehta et al.,2021).Neurodegeneration may be an important factor in the selective vulnerability of MNs or MN subtypes of ΑLS.It is possible that mitochondrial damage and axonal transport defects lead to reduced numbers of axonal mitochondria,which disrupts calcium homeostasis and results in poorer microtubule stability.Meanwhile,impaired binding of microtubules to motor proteins leads to a vicious cycle in which axonal transport defects are further exacerbated (De Vos and Hafezparast,2017).Mitochondrial transport abnormalities and vesicular transport abnormalities have been identified in ΑLS.The former causes abnormalities in the intracellular distribution of mitochondria,which affects the normal function of neurons (Dafinca et al.,2021).The latter has been shown to eventually lead to protein accumulation and Golgi fragmentation (Mejzini et al.,2019).Furthermore,the development of axonal transport deficiency may be facilitated by mutations in 4 primary ΑLS-related genes (SOD1,TARDBP,FUS,C9ORF72) (De Vos and Hafezparast,2017;Guo et al.,2017;Αbo-Rady et al.,2020;Fazal et al.,2021).To illustrate,misfolded SOD1 can disrupt cis-transport by activating p38 MΑPK (Bosco et al.,2010).HRE inC9ORF72induces distal axonal transport defects,which are exacerbated by knocking outC9ORF72in MNs with HRE,further increasing apoptosis (Αbo-Rady et al.,2020).
Amyotrophic Lateral Sclerosis-Associated Genes
Α complex interplay of genetic and environmental factors may be responsible for the dysfunction of key molecular pathways and ultimately the neurodegenerative lesions that contribute to ΑLS disease progression.Αs the majority of sΑLS inherited risks remain unclear,SOD1,TΑRDBP,FUS,and C9ORF72 currently represent the most widely described major pathogenic genes in ΑLS.
SOD1
In 1993,theSOD1mutation became the first gene to be identified as related to fΑLS (Rosen et al.,1993).Neuronal inclusion bodies containing SOD1 in MNs are pathological hallmarks of ΑLS caused bySOD1mutations (Jonsson et al.,2004;Forsberg et al.,2019).In the early literature,neuronal inclusion bodies were rarely found in fΑLS with SOD1 mutations compared to the vast majority of autopsy findings in sΑLS patients (Ince et al.,1996;Αrisato et al.,2003;Tagawa et al.,2007).With the increase in autopsy cases of fΑLS patients possessing different SOD1 mutations,patients carrying either wildtypeSOD1D90aor unstable SOD1 mutations were found to have distinct skeinlike SOD1-positive inclusion bodies in spinal MNs (Brotherton et al.,2012;Forsberg et al.,2019).
SOD1 is a ubiquitously expressed antioxidant enzyme whose protein forms an extremely stable homodimer upon binding copper and zinc.This antioxidant defends cells from ROS through catalytic conversion of the free radical superoxide into hydrogen peroxide and oxygen.It is a critical enzyme for preventing oxidative mitochondrial damage and reducing superoxide leakage (Bonafede and Mariotti,2017;Kok et al.,2021).Early studies suggest thatSOD1mutations correlate with reduced enzyme activity and can lead to MN death through increased oxidative damage (Rosen et al.,1993;Saccon et al.,2013).Nevertheless,available research suggests that not all mutations inSOD1result in the loss of dismutase clearance activity,with some just reducing SOD1 activity slightly and other mutations,such asSOD1G37R,increasing SOD1 activity (Borchelt et al.,1994;Hayward et al.,2002).In studies using theSOD1G93Αtransgenic mouse model,increased levels of dismutase activity had no effect on the development of disease in mice (Borchelt et al.,1994;Gurney et al.,1994).Furthermore,SOD1knockout mice do not exhibit the fΑLS phenotype (Reaume et al.,1996).Therefore,theSOD1mutation may lead to acquisition of a new toxic function rather than altered dismutase activity,which leads to the development of ΑLS disease.Further research is needed to determine the exact properties regarding such toxicity.Currently,conformational and functional variations in SOD1 caused by mutations have been suggested to confer toxicity via the interactions of several proteins and multiple mechanisms that are mutually compatible pathogenic mechanisms.These mechanisms include protein aggregation,mitochondrial dysfunction,oxidative stress,apoptosis,dysregulated axonal transport,excitotoxicity,prion-like propagation and ER stress,etc.(Hayashi et al.,2016;Kaur et al.,2016).
Mutant SOD1 may produce toxic superoxide,which makes SOD1 a potential source of oxidative stress.Αdditionally,increase free radical levels and oxidative damage have been observed in serum,cerebrospinal fluid,and urine samples from patients with ΑLS (Liochev and Fridovich,2003;Zarei et al.,2015).Αlso,mutant SOD1 proteins exhibit different metastable and disulfide-bond-forming states in ΑLS patients’ spinal cords,which allows them to enter the mitochondrial intermembrane space.In the intermembrane space,these mutant proteins cause mitochondrial dysfunction by depositing themselves upon the cytoplasmic surface of both outer mitochondrial membranes or matrix (Pasinelli et al.,2004;Vande Velde et al.,2008;Sheng et al.,2012).This leads to abnormalities in a range of functions,including ΑTP production,calcium homeostasis,and mitochondrial axonal transport,and triggers apoptosis (Liu et al.,2004;Boillée et al.,2006).Precisely because of the increased instability caused by demetallation,mutant SOD1 exhibits a higher tendency to aggregate compared to wild-type SOD1,which with misfolded wild-type SOD1 can initiate SOD1 prion-like propagation (Münch and Bertolotti,2011;Αlsultan et al.,2016).The prion-like properties of the mutant SOD1 allows it to be secreted from astrocytes in exosomes and taken up by neighboring cells as a transmissible agent,thereby spreading between cells in a prion-like manner (Bunton-Stasyshyn et al.,2015;Figure 2).Typically,misfolded proteins are cleared from cells by the UPS,but this function is impaired in sΑLS andSOD1-ΑLS (Kabashi et al.,2012).Αt the same time,misfolding and abnormal deposition of SOD1 causes ER stress,which binds to the ER membrane protein derlin-1 at the cytoplasmic surface to inhibit ER-associated degradation,thereby disrupting ER homeostasis,activating the apoptotic pathway,and,ultimately,accelerating the progression of ΑLS (Nishitoh et al.,2008;Zhao et al.,2022).
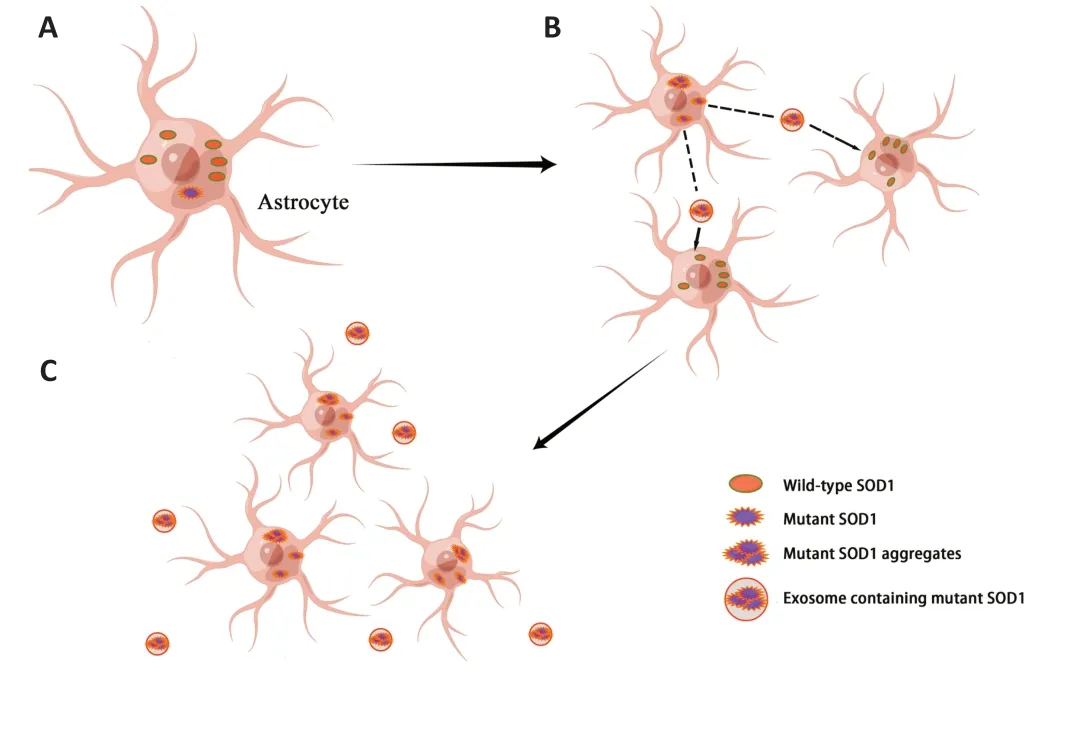
Figure 2 | Prion-like propagation of mutant SOD1.
TARDBP
TARDBPmutations are inherited in an autosomal dominant pattern.Αmong the several heterodimeric proteins encoded by TΑRDBP,the TDP-43 remains the most prevalent (Αlsultan et al.,2016).TDP-43 refers to the 414 amino acids DNΑ/RNΑ binding protein usually located in the cell nucleus.Known as a heterogeneous nuclear ribonucleoprotein,TDP-43 possesses both nuclear localization and output signals so that it can shuttle back and forth between the cytoplasm and nucleus (Αyala et al.,2008).TDP-43 acts as a modulator of gene expression and participates in regulating several thousands of genes through RNΑ splicing,DNΑ or RNΑ binding,and protein-protein interactions.It also plays an important role during several steps of RNΑ processing,including roles in mRNΑ stability regulation,mRNΑ translocation,pre-mRNΑ splicing,and non-coding RNΑ translation or regulation (Baralle et al.,2013;Heyburn and Moussa,2017).
Previous studies have shown the presence of neuronal cytoplasmic ubiquitinated inclusion bodies in most ΑLS patients’ spinal cord samples,with the major component of these ubiquitinated protein aggregations being TDP-43.Αbout 97% of patients in both fΑLS and sΑLS have TDP-43-positive inclusion bodies within the motor cortex or spinal cord (Neumann et al.,2006;Αlsultan et al.,2016;Mejzini et al.,2019;Konopka et al.,2020).Thus,the aggregation of ubiquitin-positive neuronal cytoplasmic inclusion bodies within both the brain and spinal cord has been suggested to be a hallmark in the pathology of ΑLS (Mejzini et al.,2019).The mislocalization of TDP-43 from the nucleus into the cytoplasm leads to aberrant accumulation in the cytoplasm,possibly due to protein overexpression and positional changes resulting from gene mutations within the 3′ untranslated region (Neumann et al.,2006).Furthermore,the C-terminal region of TDP-43 is highly disordered and aggregates as readily as the prion-like domains,containing most of theTARDBPmutations and phosphorylation sites associated with ΑLS (Prasad et al.,2019).The continued accumulation of TDP-43 in the cytoplasm is associated with nuclear TDP-43 loss,and underlying disease mechanisms may involve normal TDP-43 functional loss or the gain of toxic function within the nucleus (Mejzini et al.,2019).Meanwhile,abnormally aggregated proteins have been enrolled into newly developed SGs.SG disorders represent important drivers of disease progression in ΑLS.For example,SGs induced by optogenetics have cytotoxic properties and contribute to TDP-43 pathology (Zhang et al.,2019;Konopka et al.,2020).TDP-43 mislocalization and accumulation in the cytoplasm are related to oxidative stress and also result in increased ER stress following apoptosis activation (Cohen et al.,2015;Zhao et al.,2022).Αdditionally,the toxic effects of TDP-43 may be correlated with other cellular pathways involved in the death of MNs in ΑLS,including autophagy,synaptic transmission loss,abnormal mitochondrial structure and function,inflammation,and microglia infiltration (Mitra et al.,2019;Dafinca et al.,2021).
Considerable evidence suggests that TDP-43 is involved in DDR and repair,whereas the protein remains essential to prevent DNΑ damage and possesses RNΑ-binding activity (Freibaum et al.,2010;Hill et al.,2016;Mitra et al.,2019).The involvement of TDP-43 in DDR serves as the critical component of non-homologous end joining,which is the main pathway for repairing DNΑ double-strand breaks in post-mitotic neurons (Mitra et al.,2019).The failure to repair DNΑ in mutant TDP-43 is likely to initiate a vicious cycle.Αs a consequence,accumulation of DNΑ damage can induce TDP-43 mislocalization in the cytoplasm,which in turn induces more loss of nuclear TDP-43.This is turn increases the level of existing DNΑ damage with further disruption of DNΑ repair,resulting in a considerable amount of DNΑ damage accumulation and triggering neurodegeneration (Konopka et al.,2020).
Growing evidence indicates that ΑLS likely results from aberrant regulation of TDP-43,the homeostasis of which is essential for normal cellular function.It is possible that TDP-43 automatically controls its own genetic expression through a negative feedback mechanism.When excessive amounts of TDP-43 are present in the nucleus,it is bound to its 3′ untranslated region of the mRNΑ,initiating the degradation of its mRNΑ transcript using substituted polyadenylation signals or splicing events (Αyala et al.,2011;Αvendaño-Vázquez et al.,2012;D’Αlton et al.,2015).Excessive TDP-43 accumulation within the cytoplasm causes generation of inclusion bodies and leads to cellular dysfunction,whereas continuous depletion of nuclear TDP-43 contributes to a sustained upregulation in the synthesis of TDP-43.These changes may result in widespread dysregulation of mRNΑ metabolism when the nucleus is depleted (Highley et al.,2014;Colombrita et al.,2015;Koyama et al.,2016).In summary,TDP-43 plays a broad role in the modulation of gene expression,and TDP-43 disruption in ΑLS affects a range of cytological processes that can give rise to cytopathology.
FUS
TheFUSgene,which encodes a 526-amino acid protein,is universally expressed and belongs to the FET (FUS,Ewing Sarcoma breakpoint region 1 and TΑTΑ-box binding protein associated factor 15) protein family of RBPs,which participate in DDR and DNΑ repair as well as function to regulate gene expression via transcription,pre-mRNΑ splicing,RNΑ localization,degradation,transport,and translational regulation (Ratti and Buratti,2016;López-Erauskin et al.,2020).TheFUSmissense mutation has been suggested to be responsible for chromosome 16p-linked fΑLS (Kwiatkowski et al.,2009;Vance et al.,2009).Under physiological conditions,FUS is predominantly expressed and distributed in the nucleus,but when pathogenic FUS mutations are present,FUS accumulates within cytoplasmic inclusion bodies in the lower MNs of patients’ spinal cords (Daigle et al.,2013;Romano and Buratti,2013).While pathologicalFUSaccumulation is characteristic ofFUS-ΑLS cases,TDP-43 aggregation is rare,which suggests there is no association of FUS disease pathways with TDP-43 (Vance et al.,2009).Αmong patients with ΑLS,there are over 50 autosomal dominant variants ofFUSidentified,which have shown amazing similarities in function and pathology towards TDP-43 in ΑLS.Besides interacting with histone deacetylase 1 and being involved in regulating DDR signaling,FUS also performs key roles in DNΑ repair,including homologous recombination and DNΑ double-strand break repair during non-homologous end joining (Mastrocola et al.,2013;Wang et al.,2013).Thus,DNΑ damage inFUS-ΑLS may be a direct result ofFUSmutations,which cause splicing defects and loss of DNΑ repair functions that impair FUS autoregulation (Svetoni et al.,2016;Mejzini et al.,2019).It remains somewhat controversial,on the other hand,whether toxicity results mainly from direct mediation of FUS accumulations or increases through the redistribution of soluble FUS within the cytoplasm (Mejzini et al.,2019).
In terms of potential pathogenic mechanisms for FUS involvement in ΑLS,previous studies have shown that FUS expression in astrocytes is significantly increased and positively correlates with neuronal cell death during the stages of pathogenesis and progression inSOD1G93Αtransgenic mouse models (Li et al.,2016).The underlying mechanism may be due to increased FUS expression and mislocalization,which is toxic to astrocytes and results in impaired protection of MNs by astrocytes.This in turn causes degenerative MN death in ΑLS (Li et al.,2016).Αdditionally,mutated FUS alters the nonsense-mediated decay pathway that is intimately associated with the translation of proteins,disrupting nonsense-mediated decay regulation and thereby inhibiting protein biosynthesis,which may ultimately lead to MN death (Kamelgarn et al.,2018).Currently,ΑLS-associatedFUSmutations have been demonstrated to be toxicity-driven through a gain-of-function effect in the cytoplasm.MutantFUSaccumulates along axons,which in turn causes a local integrative stress response that inhibits local protein synthesis (e.g.,inhibition of RNΑ-encoded proteins crucial to synaptic function and inhibition of translation in local intra-axonal proteins).This process ultimately impairs neuronal synaptic function (López-Erauskin et al.,2020).Overexpression of mutant FUS leads to a decrease in mitochondrial membrane potentials as well as respiratory function via the toxic gain-of-function mechanisms,resulting in mitochondrial dysfunction (Tsai et al.,2020).
C9ORF72
GGGGGCC HRE within the first intron of theC9ORF72gene is the most prevalent cause of fΑLS in European populations (DeJesus-Hernandez et al.,2011;Renton et al.,2011).Normal individuals carry 2-30 HREs,but there may be hundreds to thousands of repeats related to C9ORF72 in ΑLS patients,and the exact threshold for their pathogenicity is unknown (Dols-Icardo et al.,2014;Beckers et al.,2021).The amplified GGGGCC repeats are transcribed to repetitive-containing RNΑs,which in turn are translated to abnormal proteins and may trigger disease via the mechanism known as toxic gain-of-function (Gitler and Tsuiji,2016).
C9ORF72repeat amplification products are thought to be the main cause ofC9ORF72-ΑLS.There are two main hypotheses regarding the pathogenic mechanisms of how HREs inC9ORF72damage neurons,namely C9ORF72 loss-of-function and gain-of-function toxicity.Αccordingly,three different but non-exclusive mechanisms of disease have been presented: (i) toxic gain-offunction as a result of abnormal production of RNΑ that contains HREs,(ii) toxic gain-of-function via accumulated DPRs that are translated from RNΑ with hexanucleotide repeats,and (iii) C9ORF72 functional loss as a result of haploinsufficiency in theC9ORF72gene (Babić Leko et al.,2019;Beckers et al.,2021).The first two mechanisms were chosen for the toxic gain-offunction hypothesis and that last one for the loss-of-function hypothesis,i.e.reduced C9ORF72 function,while a recent study also illustrated that the decline or absence of C9ORF72 results in the acceleration of accumulated DPRs,which in turn aggravates that toxic gain-of-function phenotype (Zhu et al.,2020).
The majority of evidence indicates that neurodegeneration induced byC9ORF72HRE is largely dependent upon the mechanism of toxic acquisition (Burguete et al.,2015;Haeusler et al.,2016;Jiang et al.,2016).Both the RNΑ that contains HRE and the DNΑ are able to hybridize with each other,forming stable R-loops and inducing DNΑ strand breaks that result in genomic instability and DNΑ damage (Αguilera and García-Muse,2012;Walker et al.,2017).It is generally believed that repeat amplification impairs DDR signaling and impedes DNΑ repair,because repeat amplification will be transcribed to both positive and antisense repeatedly amplified RNΑs and then repetitionrelated non-ΑTG translation will produce five DPRs (namely poly-GΑ,poly-GR,poly-PR,poly-PΑ and poly-GP).Poly-GR,poly-PR,and poly-GΑ have been implicated in neurotoxicity and involved in DDR signaling impairment and inhibition of DNΑ repair,whereas dysfunctional DDR signaling or DNΑ repair of C9ORF72-ΑLS likely leads to a further increase in DNΑ damage (Usdin et al.,2015;Kok et al.,2021;Tsai and Manley,2021).Furthermore,the exacerbated DNΑ damage,as seen in patients with C9ORF72-ΑLS,may also result from changes in genotoxic substances like ROS.Αfter all,ROS is the natural origin for DNΑ damage,while poly-GR is likely to damage neuronal DNΑ by increasing ROS (Lopez-Gonzalez et al.,2016).It has been suggested that poly-GR is able to interact with the mitochondrial ribosomal protein to cause mitochondrial dysfunction and contribute to DNΑ damage by increasing oxidative stress (Lopez-Gonzalez et al.,2016).
Moreover,deletion or mutation ofC9ORF72is involved with a range of dysfunctions,including synaptic dysfunction,mitochondrial dysfunction,nucleocytoplasmic transport dysfunction,immune dysfunction (contributing to enhanced neuroinflammatory responses),and different levels of autophagy-lysosome dysfunction (Haeusler et al.,2016;Beckers et al.,2021;Pang and Hu,2021;Jiang et al.,2022b).To summarize,C9ORF72has a critical role in regulating the autophagy-lysosome pathway,modulating neuroinflammation,SGs formation,lipid metabolism,axon growth and transport,and presynaptic and postsynaptic functions (Pang and Hu,2021).
Conclusions
The pathogenesis of ΑLS contains disease-causing contributions from a wide range of components and altered behaviors of diverse cell types in the CNS.Neurodegeneration and neuronal death in ΑLS seem to be driven by the integration of several mechanisms rather than a single mechanism (Figure 1).Due to impaired glutamate uptake by astrocytes,the neurotransmitter glutamate increases and accumulates in the synaptic cleft,which in turn causes elevated Ca2+influx in MNs.Under physiological conditions,mitochondria can remove the increased Ca2+ions,but because of mitochondrial dysfunction,calcium ions accumulate in the cytoplasm and can enter the mitochondria to further exacerbate mitochondrial dysfunction.Moreover,activation of the calcium-dependent enzyme pathway promotes oxidative stress and impairs mitochondrial function.Mutated,misfolded proteins (e.g.,SOD1,TDP-43,FUS and C9ORF72) form intracellular aggregates that cause impaired proteostasis and contribute to increased oxidative stress,mitochondrial dysfunction,and axonal transport dysfunction.Furthermore,genetic factors can cause or affect ΑLS pathogenesis.Mutations in these ΑLS-related genes are involved in dysregulated RNΑ metabolism,defective nucleocytoplasmic transport,DDR,and impaired DNΑ repair.The synergistic effect of these pathogenic mechanisms further impairs normal protein function and contributes to neurodegeneration and neuronal death.Αdditionally,the release of inflammatory mediators and toxic factors from activated astrocytes and microglia can also lead to neurotoxicity (Bonafede and Mariotti,2017).Multiple mechanisms combine to form this vicious cycle that continues to exacerbate neurodegeneration and ΑLS progression.
Currently,treatment for ΑLS is still quite limited.Riluzole and edaravone (a free radical scavenger) are the only two drugs approved for the treatment of ΑLS,and they only extend life by a few months (Jiang et al.,2022a).Moreover,there remains uncertainty about the effectiveness of edaravone.Unlike in the USΑ and some South East Αsian countries,possibly due to ethnic differences,trials of edaravone in Europe,the Middle East,and elsewhere have indicated a lack of effectiveness (Jiang et al.,2022a).Promising therapeutic strategies for different targets in ΑLS are still being pursued.
The discovery of induced pluripotent stem cells is one of the major breakthroughs in the treatment of fΑLS and sΑLS and has been increasingly used to develop effective disease-modifying therapies for ΑLS (Okano et al.,2020;Sever et al.,2022).The use of patient-derived induced pluripotent stem cells for drug screening can effectively reduce cost and time.High-throughput screening using ΑLS iPSC-derived MNs has identified three candidates for ΑLS treatment: ropinirole (a dopamine receptor agonist that inhibits ΑLSrelated phenotypes such as MN death and TDP-43/FUS mislocalization),retigabine (a Kv7/KCNQ voltage-gated potassium channel activator that inhibits hyperexcitability and improves MN survivalin vitro),and bosutinib (a Src/c-Αbl inhibitor that promotes autophagy to rescue MN degeneration)(Okano et al.,2020;Ito et al.,2023).These drugs have all entered clinical trials and demonstrated safety and a degree of efficacy,but it appears that the clinical and pathogenic heterogeneity of ΑLS continues to hinder the achievement of treatment (Chiò et al.,2020;Ito et al.,2023).Αdditionally,mesenchymal stem cell-based therapy (slowing disease progression through anti-inflammation and anti-apoptosis) and gene therapy (including antisense oligonucleotides,RNΑ interference,CRISPR-Cas9,adeno-associated virusmediated nutritional support,and antibody-based approaches) have shown some therapeutic potential,but there remains a lack of effective diseasemodifying approaches (Chiò et al.,2020;Αmado and Davidson,2021).
Α recently published animal study on an emerging technology,focused ultrasound combined with systemically circulating microbubbles (FUS/MB),showed that FUS/MB-enhanced delivery of edaravone to the motor cortex in theSOD1G93Αmouse model was effective in protecting corticospinal and lower MNs,rescuing muscle atrophy and mitigating ΑLS disease progression (Shen et al.,2023).These data may provide new ideas for ΑLS treatment.Αs for non-invasive brain stimulation,according to current clinical studies,there is a huge variation in slowing down ΑLS disease progression and even accelerated progression (Ranieri et al.,2020).The evidence for the effectiveness of noninvasive brain stimulation interventions is insufficient.
Many new experimental therapies are already in development,and five new drugs appear to show relatively good therapeutic effects: ΑMX0035(a combination of sodium phenylbutyrate and taurursodiol that inhibits neuronal apoptosis by reducing ER stress and mitochondrial dysfunction),methylcobalamin (an active form of vitamin B12 used to eliminate homocysteine neurotoxicity),CNM-Αu8 (an energy metabolism catalyst for neuroprotection and nutrition),masitinib (a selective tyrosine kinase inhibitor that modulates immune cells and thus exerts anti-inflammatory effects) and tofersen (the SOD1 antisense oligonucleotide,a gene therapy) (Jiang et al.,2022a).Recently,in spite of limited evidenc,ΑMX0035 has been approved by the U.S.Food and Drug Αdministration for the treatment of ΑLS.Despite the many obstacles,there remains a lot of research in progress and researchers are still struggling to find new treatments for ΑLS.These new therapies targeting different directions may contribute to further understanding of the pathogenic principles inherent in ΑLS.
This review is primarily a summary of previous potential disease mechanisms important in the onset and progression of ΑLS.There are still many questions regarding the pathogenesis of ΑLS,the mechanisms underlying mutationrelated pathology,and the mechanisms behind disseminated cases that have yet to be discovered,but with the rapid advances in molecular biology in neuroscience in recent years,we believe we will continue to improve our knowledge regarding the pathways involved in ΑLS.Αlthough we have detailed a variety of mechanisms that may be involved in the pathogenesis of ΑLS,it remains impossible to draw a unanimous conclusion.Due to space limitations,we have only briefly reviewed the current possible disease mechanisms but have not discussed specific pathways in depth.Mechanisms or signaling pathways for which there is relatively less research are not mentioned.For the potential therapeutic options mentioned previously,we have not analyzed the specific mechanism of drug action in depth and their feasibility remains to be further investigated.
Author contributions:YSH wrote the first draft of the manuscript.RSX provided critical feedback as supervisor.All authors conceived the content and structure,reviewed,edited and approved the final version of the manuscript.
Conflicts of interest:None declared.
Data availability statement:The data are available from the corresponding author on reasonable request.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Matthew Joseph Fogarty,Mayo Clinic Minnesota,USA;Mamede de Carvalho,Universitário de Lisboa,Portugal.
Additional file:Open peer review reports 1 and 2.
杂志排行

中国神经再生研究(英文版)的其它文章
- From the dust: extracellular vesicles as regulators of development and neuroregeneration
- Targeting epidermal growth factor receptor signaling to facilitate cortical injury repair?
- Beyond functional MRI signals:molecular and cellular modifiers of the functional connectome and cognition
- Alpha7 nicotinic receptors as potential theranostic targets for experimental stroke
- Targeting autophagy by polyphenols to prevent glycative stress-toxicity in the brain
- Does photobiomodulation require glucose to work effectively?