颗粒Co3O4-g-C3N4吸附剂的制备及其对甲基橙的吸附性能研究
2024-02-13熊波黄海茵黄卓杰刘长宇徐晓龙
*熊波 黄海茵 黄卓杰 刘长宇 徐晓龙
(五邑大学生物科技与大健康学院 广东 520090)
有机染料是水环境污染物之一,对水生生物和人体有害。去除水中污染物的方法有膜分离、光催化降解和吸附法等。吸附法由于成本低、方法简便,在污水处理方面得到了广泛应用。
石墨氮化碳(g-C3N4)是一种新型的以碳氮键取代碳碳键的二维非金属聚合物,可作为吸附剂[1]。g-C3N4的基本结构是sp2杂化C和N原子的类石墨平面,具有富缺陷的表面以及富电子的氮原子官能团,有利于通过亲核相互作用增强其吸附性能,为废水中有毒物质的吸附提供活性位点[1]。张守伟课题组[2]通过热聚合-刻蚀法制备三维大孔g-C3N4用于吸附U(Ⅳ);Xie等人[3]通过热聚合法制备ATP/g-C3N4用于吸附Cd(Ⅱ);Wang等人[4]通过水热法制备g-C3N4/MnFe2O4/Fe-MIL(100)复合吸附剂用于吸附环丙沙星、土霉素和吲哚美辛;Kaur等人[5]制备g-C3N4@NiCoLDH复合材料用于吸附水杨酸和亚甲基蓝。
本文在双氰胺(Dicyandiamide,DICY)和氯化铵(NH4Cl)热解研究的基础上,在原料中加入六水硝酸钴(Co(NO)2·6H2O)通过一步热聚合法合成Co3O4-g-C3N4,表征了Co3O4-g-C3N4的形貌、晶相结构、元素价态分布以及孔径分布等,同时对其吸附的动力学与热力学模型进行研究。结果表明,钴元素的引入改变了g-C3N4的孔隙结构、使其比表面积增大、吸附活性位点增多,从而增强其吸附能力。
1.实验分析
(1)仪器及作用分析
实验中用到的主要设备有箱式马弗炉(合肥科晶材料技术有限公司,KSL-1200X-M(27L)型)、紫外可见分光光度计(日本岛津制作所,UV-1900型)和磁力搅拌器(德国海道尔夫公司,Hei-Tec型)。材料表征用到了扫描电子显微镜(捷克泰思肯公司,TESCAN MIRA4型)、X射线衍射仪(日本理学Rigaku公司,Rigaku SmartLab 9kW型)、全自动比表面积及孔隙度分析仪(美国麦克默瑞提克公司,ASAP 2460型)和X射线光电子能谱仪(美国赛默飞世尔科技公司,Thermo Scientific K-Alpha型)。
(2)主要试剂
Co(NO)2·6H2O(麦克林,分析纯);NH4Cl(阿拉丁,分析纯);DICY、甲基橙、无水磷酸二氢钠(上海笛柏生物科技有限公司,分析纯);磷酸二氢钾(上海穗试公司,分析纯)和氢氧化钠(广东光华科技股份有限公司,分析纯);实验用水为超纯水(>18.2MΩ·cm)。
(3)吸附剂制备
制备g-C3N4吸附剂:将各为0.8000g的DICY和NH4Cl混合并研磨均匀,以5℃/min的升温速率加热至550℃并恒温3h。自然冷却至室温后将样品从炉中取出并研磨成粉末,即为g-C3N4[6]。
制备Co3O4-g-C3N4吸附剂:将DICY、Co(NO)2·6H2O和NH4Cl按一定质量比例混合并研磨均匀。将混合物放入箱式马弗炉,以5℃/min的速率梯度升温至特定的目标温度并维持恒温退火特定时长,待自然冷却至室温后,将样品从炉中取出并研磨成粉末,即为Co3O4-g-C3N4材料。在实验中,DICY和Co(NO)2·6H2O的总质量为1.000g,以不同的质量比17:3、3:1和4:1,退火温度500℃、550℃和600℃,恒温时间2h、3h和4h等对其制备条件进行优化。
(4)吸附剂表征
扫描电镜(SEM)使用肖特基场发射电子枪,样品表面喷涂铂金,放大倍率为2万~10万倍,加速电压为200~300keV。X射线衍射(XRD)晶相结构表征中,采用扫描速度10°/min,角度范围10°~80°。比表面积测试(BET)在77.3K以N2为吸附质,在200℃温度下脱气6h。X射线光电子能谱(XPS)激发光束的束斑大小为400μm,实验中使用的工作电压为12kV,灯丝电流为6mA,在进行窄谱扫描时,通能设置为50eV。
(5)吸附性能研究
吸附实验:将一定质量的Co3O4-g-C3N4吸附材料分散在甲基橙(MO)溶液中进行吸附反应(磁力搅拌,n=1400r/min),每隔6min取一定量的MO溶液,经过滤后用紫外可见分光光度计进行表征。吸附效率和吸附容量分别按公式(1)、公式(2)计算。
式中,At为MO溶液被吸附既定时间t(min)后溶液的吸光度;A0为MO溶液的初始吸光度。
式中,m、V、ρ0和ρt分别为吸附剂的投加量(mg)、MO溶液体积(mL)、初始浓度(mg/L)和吸附既定时间t(min)时的浓度(mg/L);Qt为吸附时间t对应的吸附容量(mg/g)。
吸附-脱附实验:将一定质量的吸附剂(如0.2000 g)投入2.0L MO溶液(50mg/L)中吸附1h(n=1400 r/min),静置10min后离心分离,过滤物在60℃烘箱中烘干3h,取出自然冷却至室温后研磨至粉末状。称取一定质量该粉末(如30mg)分散在不同pH的磷酸缓冲溶液中搅拌10min(n=1400r/min),用紫外可见分光光度计表征上清液。
2.结果与讨论
(1)材料表征
①微观形貌表征
DICY与NH4Cl加热聚合得到的g-C3N4是紧密堆叠在一起的厚层多孔块体且分布不规则的厚块状材料(图1A)。在原料中加入Co(NO)2·6H2O后,材料呈马蜂窝状形貌,颗粒变小,粒度分布较均匀(图1B)。对比二者,在原有的前驱体中加入Co(NO)2·6H2O后,所制备的材料形貌明显发生变化,这可能会导致比表面积增大,吸附活性位点增多,从而增强吸附能力。

图1 SEM图像
②XRD分析
g-C3N4、Co3O4-g-C3N4的XRD图谱如图2所示。在27.25°(002晶面)和13.03°(001晶面)处分别观察到两个特征衍射峰(JCPDS 87-1526)。(002)平面归因于共轭三嗪芳香族薄片的层间叠加反射,表明石墨化的形成;(001)平面归因于七嗪单元平面内结构填充基序。引入Co元素以后,观察不到13.03°处(001)晶面,27.25°处(002)晶面尖峰变得较平稳,说明石墨相氮化碳七嗪单元(001)平面结构、共轭三嗪环芳香族的层间叠加反射(002)晶面结构在引入钴元素时遭到破坏。经过高温聚合后材料的XRD中,除了有石墨化碳峰以外,在36.86°、44.58°、55.82°处出现了小峰,对应于Co3O4的311、400、511晶面(JCPDS card no.74-2120),说明材料中有Co3O4存在[7-9]。通过观察XRD峰型,可以认为Co3O4与g-C3N4形成有效复合。
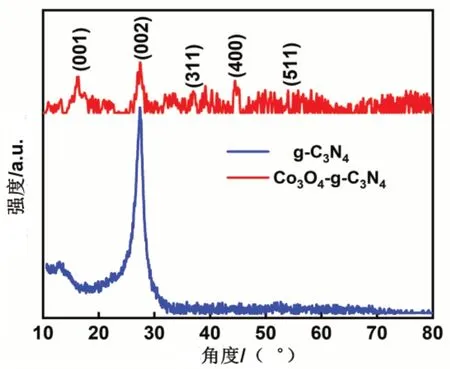
图2 g-C3N4和Co3O4-g-C3N4的XRD图谱
③XPS分析
通过XPS分析得到吸附剂材料的结果,如图3所示。图3A中包含4组结合能峰,对应于材料中的C、N、O和Co元素。使用Advantage处理数据后,可以确定该材料中各元素含量分别为40.73%的C、28.38%的O、21.66%的N和9.23%的Co。图3B中,284.38eV和287.88eV是XPS光谱图中碳的两个峰值,对应于g-C3N4材料中不同类型的碳化合物。具体来说,284.38eV峰值代表sp2杂化的C-C单键或C-C双键,这些键通常存在于类似石墨烯的平面结构中。而287.88eV峰值代表含N芳香环(N-C-N)中的sp键碳,在g-C3N4中,这些类型的碳占据着主要的碳种类[7-8]。图3C中398.58eV的峰可归属于三个含氮基团的N原子,分别是398.40eV、399.17eV和400.07eV。具体而言,398.40eV对应于吡啶环中的N原子(C-N-C键),399.17eV对应于与钴原子配位的N原子(Co-Nx),而400.07eV对应于吡啶环上的N原子(N-(C)3)[9]。图3D中在531.85eV处的O1s峰,表明g-C3N4中存在结合C原子的氧官能团(如COOH和OH)[8]。图3E为Co 2p能谱图,图中797.33eV和781.77eV的两个峰与Co3O4的Co 2p1/2和Co 2p3/2峰一致,揭示了Co2+和Co3+的存在,表明所得异质结中Co的主要形式为Co3O4。786.10eV、803.37eV这两个峰是Co 2p中的典型卫星峰。结合XRD结果来看,可以将其吸附剂命名为Co3O4-g-C3N4[9]。
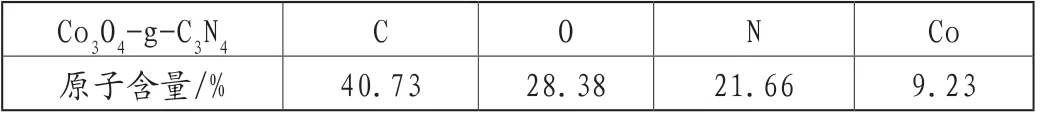
表1 用Advantange处理XPS数据可知各元素的含量
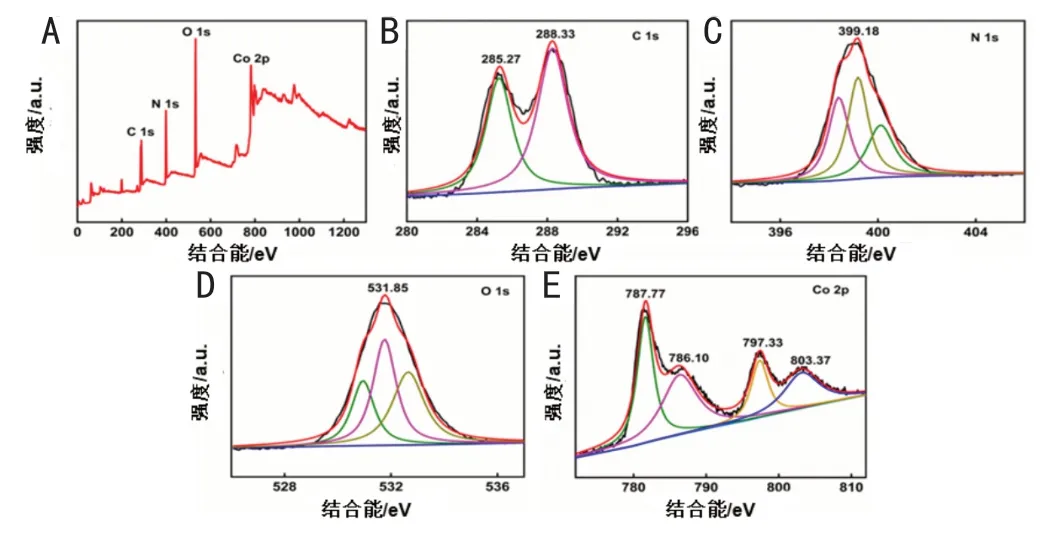
图3(A)Co3O4 -g-C3N4、(B)C 1s、(C)N 1s、(D)O 1s和(E)Co 2p的XPS光谱
④BET分析
从图4数据分析可知,氮气吸附-脱附等温线具有明显的迟滞环,这表明Co3O4-g-C3N4复合材料中存在介孔结构[10]。从孔径分布图可以看出,该催化剂拥有丰富的介孔结构,主要孔径大小分别为2.60nm、3.60nm和30.40nm三种,且大多数孔的孔径为3.60或者30.40nm,2.60nm的孔较少。吸附剂的BET比表面积为47.82m2/g,总孔隙体积为0.19cm3/g,平均孔径为16.24nm。Co3O4-g-C3N4的回滞环较大,说明其孔径类型较多,与孔径分布图一致。文献[6]中以双氰胺为前驱体制备的g-C3N4,比表面积为2.90m2/g,Co3O4-g-C3N4吸附剂与之相比,比表面积提高15.49倍。这说明在制备过程中引入Co(NO)2·6H2O后,材料的比表面积显著增大,活性位点也增多,一定程度上会使吸附能力增强。
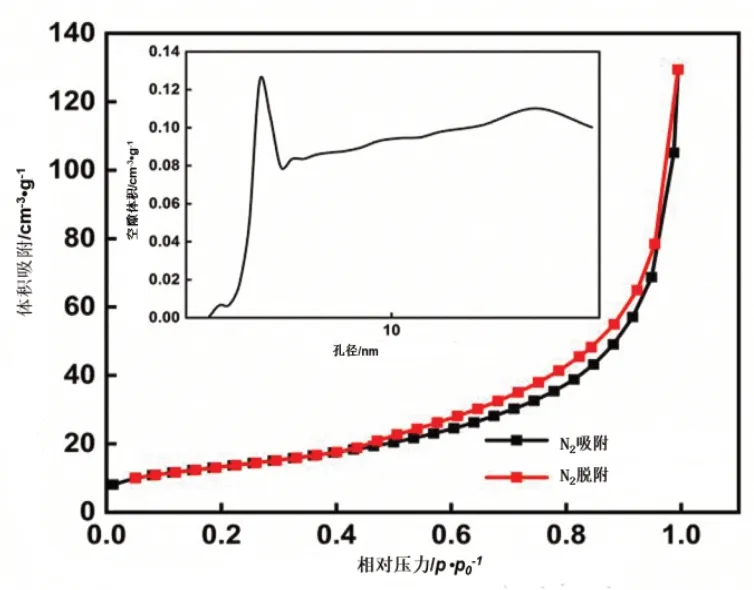
图4 Co3O4-g-C3N4的氮吸附-解吸等温线和孔径分布曲线
(2)吸附材料制备条件的优化
文献[11]报道三聚氰胺(C3H6N6)和DICY为前驱体制备g-C3N4的产率较高,可分别达到40.5%和31.1%;而硫氰酸铵(NH4SCN)、硫脲(CH4N2S)的产率仅有9.21%、7.25%。本文选择产率相对较高的DICY为氮、碳源前体,经热聚合后,钴掺杂石墨相氮化碳的吸附效率是本体的3.24倍(图5A)。在500℃、550℃、600℃时制备的吸附剂,吸附效率分别为17.42%、94.27%、7.00%,550℃时制备的材料吸附能力最好(图5B),以此为吸附剂的优选聚合温度。以吸附效率为判断依据优化前驱体的最佳质量比为4:4:1(DICY:NH4Cl:Co(NO)2·6H2O,图5C)。该材料在2h、3h、4h聚合时长所对应的吸附效率分别为57.24%、94.27%、66.67%(图5D),故选择3h为该材料的最佳聚合时长。综上,在前驱体DICY与Co(NO)2·6H2O质量比为4:1时在550℃恒温3h,制备出的材料吸附容量最大。
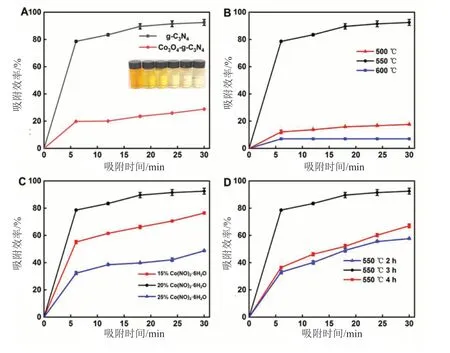
图5 不同(A)吸附剂(插图A)、(B)聚合温度、(C)Co(NO)2·6H2O用量和(D)550℃聚合持续时间的吸附效率(w%为前体中Co(NO)2·6H2O的质量分数,30mg吸附剂加入100mL 30mg/L MO溶液中,1400r/min磁力搅拌)
(3)吸附效率的影响因素
接下来,对吸附剂投加量、甲基橙初始浓度、搅拌速度、吸附时长等吸附条件进行考查,得出以下主要结论:吸附剂投加量为20mg、30mg时,30min吸附效率分别为77.26%、94.27%;而当增加吸附剂的用量到40mg时,吸附效率可在12min内达到95.14%,这表明增加吸附剂的用量可以提升吸附速率,显著缩短吸附时间(图6A)。综合考虑吸附效率和吸附时间,选取吸附剂投加量为30mg做接下来的实验。随着MO浓度的增加,吸附剂的吸附效率逐渐变低。在MO浓度为20mg/L、30mg/L、40mg/L、50mg/L,30min时吸附效率分别为94.27%、85.53%、77.86%、66.32%(图6B)。另外,吸附剂在水中传质速度也是影响吸附效率的重要因素。分别选取600r/min、1000r/min、1400r/min三个搅拌速度,对应吸附效率分别为84.57%、89.76%、94.27%(图6C)。因此,选择了搅拌器的最高转速1400r/min为该吸附实验的搅拌速度。最后,对吸附时间进行探讨,向50mg/LMO溶液中投加30mg吸附剂,1h后吸附效率为71.00%(图6D),而吸附30min后吸附效率为66.32%(图6B),吸附效率相差不大,吸附30min就几乎能够达到吸附饱和,继续增加时间对提高其吸附效率影响较小。在以上最优条件下,可得到该材料的最大平衡吸附容量为118.83mg/g。与近期相关文献[12-15]中的材料相比,从最大平衡吸附容量单一参数来看,Co3O4-g-C3N4是Magnetite-CTAB的4.26倍,略高于文献报道的Fe-La/MgO、Activated Pine Cone biochars、Activated Carbon材料(表2)。Co3O4-g-C3N4吸附剂的良好吸附能力可以归因于以下三个因素:①g-C3N4是一种二维材料,具有丰富的缺陷位和官能团,这些缺陷位和官能团可以提供足够的吸附活性位点来吸附染料分子。②引入Co3O4扩展了g-C3N4的π共轭体系,通过π-π堆积作用,促进Co3O4-g-C3N4吸附剂对MO的吸附。③Co3O4-g-C3N4吸附剂比表面积相比于本体g-C3N4增加了15.49倍,活性位点显著增多,使吸附能力增强。
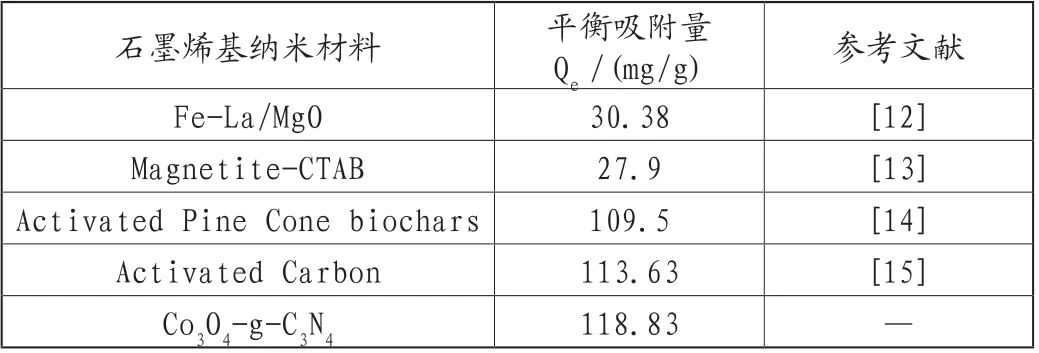
表2 本工作与近期相关文献中材料的平衡吸附容量比较

图6 不同(A)吸附剂用量、(B)MO浓度、(C)搅拌速率和(D)吸附时间下的吸附效率(除非另有说明,将30mg吸附剂加入100ml 20mg/L的MO溶液中,磁力搅拌1400r/min)
(4)动力学、热力学分析
吸附动力学是通过实验测量吸附容量随时间变化的曲线,并通过拟合得到吸附动力学参数。如图7A所示,在pH值为6.0的条件下,将30mg的吸附剂加入到浓度为20mg/L的MO溶液中(磁力搅拌,n=1400r/min),在接触时间分别为6min、12min、18min、24min和30min时,吸附容量分别为52.67mg/g、56.07mg/g、60.57mg/g、62.16mg/g和62.36mg/g。对数据进行拟合分析表明,在Co3O4-g-C3N4上甲基橙的吸附符合准二阶动力学模型(表3,相关系数r=0.999)。
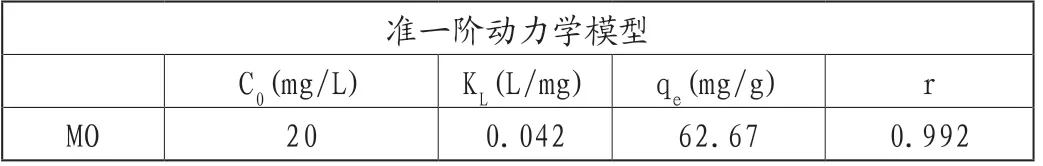
表3 Co3O4-g-C3N4吸附剂在MO溶液中吸附动力学的拟合结果
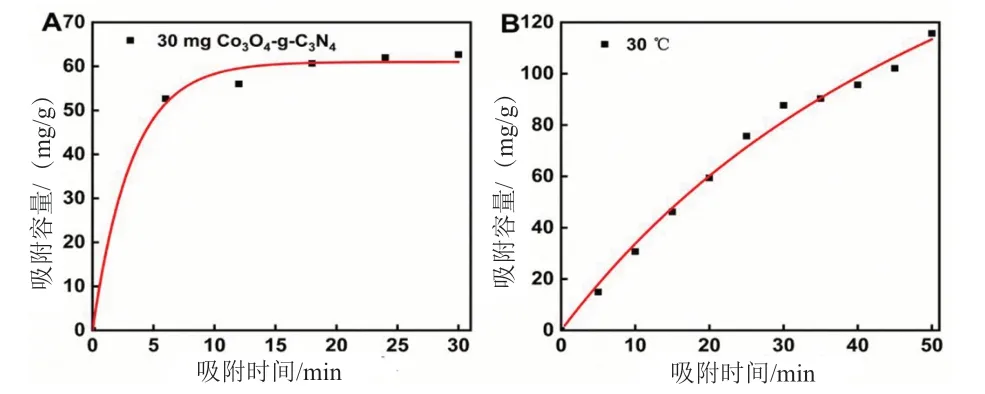
图7 MO在Co3O4-g-C3N4吸收剂上的吸附动力学(A)和等温线(B)((A)在100mL 20mg/L MO溶液中加入30mg吸附剂,pH=6.0,磁力搅拌1400r/min;(B)在100mL MO溶液中加入30mg吸附剂,pH=6.0,180r/min摇动)
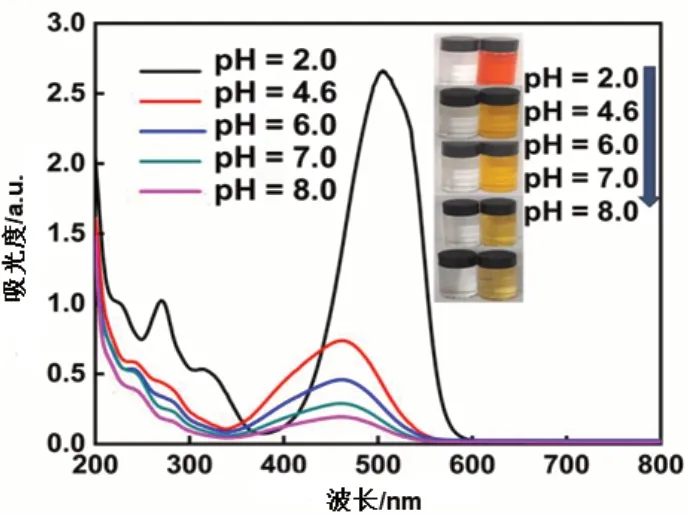
图8 不同pH下MO解吸的紫外-可见光谱和照片(30mg吸附饱和的吸附剂,加入100mL PBS,磁搅拌速度1400r/min)
用MO溶液浓度和各浓度的平衡吸附容量可分析吸附等温模型。吸附等温线如图7B所示,在规定条件下测得各浓度的吸附容量(qe)分别为14.93mg/g、30.70mg/g、46.13mg/g、59.40mg/g、75.67mg/g、87.7mg/g、90.33mg/g、95.67mg/g、102.07mg/g、115.70mg/g。数据通过拟合分析可知,在Co3O4-g-C3N4上甲基橙的吸附符合Langmuir吸附等温模型(表4,相关系数r=0.990)。
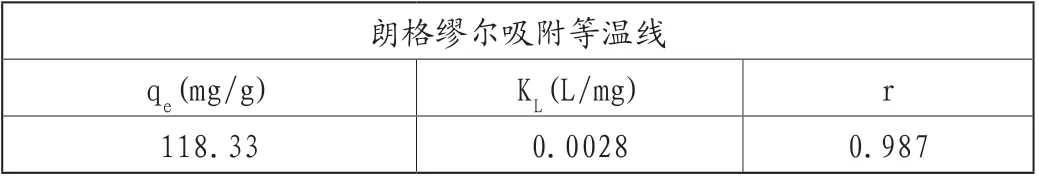
表4 Co3O4-g-C3N4吸附剂在MO溶液中吸附热力学的拟合结果
(5)吸附-脱附实验
向四种不同pH值的缓冲溶液中投加30mg吸附饱和的吸附剂并进行10min的脱附实验后,发现pH=4.6条件下脱附效果较好,说明该材料容易在酸性条件下脱附。在pH=2.0的缓冲溶液中脱附时,脱附出来的甲基橙浓度为43.41mg/L。此时,甲基橙的最大吸收波长发生红移,从461nm变为495nm。这是因为甲基橙本身是一种酸碱指示剂,在酸性条件下,甲基橙分子发生质子化反应,在pH≤3.10时呈现红色。总体而言,无论在酸性、中性或碱性条件下,通过紫外可见吸收光谱表征以及脱附前后样品瓶的对比,都明确地表明甲基橙能够被脱附出来。
3.结论
本文采用一步热聚合法制备Co3O4-g-C3N4吸附剂材料。该材料呈颗粒状,比表面积为47.82m2/g,总孔隙体积为0.19cm3/g,平均孔径为16.24nm,对含有苯环结构的甲基橙表现出较好的吸附性能,最大平衡吸附容量为118.83mg/g,优良的吸附性能可主要归因于引入Co3O4后材料比表面积增大,活性位点增多。
