Ferroptosis mechanism and Alzheimer’s disease
2024-02-11LinaFengJingyiSunLingXiaQiangShiYajunHouLiliZhangMingquanLiCundongFanBaoliangSun
Lina Feng, Jingyi Sun, Ling Xia, Qiang Shi, Yajun Hou, Lili Zhang, Mingquan Li, Cundong Fan,*, Baoliang Sun,*
Abstract Regulated cell death is a genetically determined form of programmed cell death that commonly occurs during the development of living organisms.This process plays a crucial role in modulating homeostasis and is evolutionarily conserved across a diverse range of living organisms.Ferroptosis is a classic regulatory mode of cell death.Extensive studies of regulatory cell death in Alzheimer’s disease have yielded increasing evidence that ferroptosis is closely related to the occurrence, development,and prognosis of Alzheimer’s disease.This review summarizes the molecular mechanisms of ferroptosis and recent research advances in the role of ferroptosis in Alzheimer’s disease.Our findings are expected to serve as a theoretical and experimental foundation for clinical research and targeted therapy for Alzheimer’s disease.
Key Words: Alzheimer’s disease; apolipoprotein E; Fe2+; ferroptosis; glial cell; glutathione peroxidase 4;imbalance in iron homeostasis; lipid peroxidation; regulated cell death; system Xc–
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder that affects the central nervous system and is characterized by a gradual decline in memory,logical reasoning, and language function (Ballard et al., 2011).The increasing prevalence of AD in the aging population has resulted in this condition becoming recognized as a considerable medical and social challenge for humanity.According to an epidemiological survey carried out in 2020, there were approximately 15.07 million patients with dementia over the age of 60 years in China, including 9.83 million patients with AD (Jia et al., 2020).Currently, China has the largest number of people with dementia in the world(Chinese Society of Dementia and Cognitive Impairment, 2022).Research has projected that by 2050, the number of patients with AD in China will reach 25 million; this has important implications for both individuals and society(Chen and Han, 2018).However, the pathogenesis of AD has yet to be fully elucidated, and there is an urgent need to develop drugs that can completely cure or alleviate the symptoms of AD.The main pathological features of AD are neuritic plaques, which are formed by the deposition of extra-neuronal amyloid-β (Aβ), and neurofibrillary tangles (NFTs), which are formed by the abnormal aggregation of hyperphosphorylated tau protein within neurons(Gerrits et al., 2021).Although numerous drugs can target these pathological features, these drugs do not cure or alleviate the symptoms of AD.Therefore,there is a critical need to further investigate the pathological factors that induce AD.
Cell death is a vital biological process that is irreversible and plays a crucial role in maintaining normal development and homeostasis in the body (Fuchs and Steller, 2011).Regulated cell death (RCD) and accidental cell death are two very different models of cell death.RCD is a genetically determined form of programmed cell death that encompasses a variety of types, including apoptosis, necroptosis, autophagy, ferroptosis, pyroptosis, and cuprotosis(Green, 2019; Bedoui et al., 2020; Tsvetkov et al., 2022).Ferroptosis is a regulatory mode of cell death that was only discovered recently.Ferroptosis is a disorder of iron metabolism and lipid peroxidation (LPO);the main molecular mechanism involved is an imbalance between oxidative damage and antioxidant defense (Riegman et al., 2020; Chen et al., 2022).Research has shown that ferroptosis is characterized by disturbances in iron metabolism, the accumulation of reactive oxygen species (ROS), reduced levels of glutathione (GSH), and the inactivation of glutathione peroxidase 4 (GPX4) (Huang et al., 2020).These events are also important pathological factors for AD and cognitive dysfunction.Therefore, in this review, we aimed to describe the molecular mechanisms underlying ferroptosis and the recent research advances in AD.Our findings are expected to provide a new direction for clinical research and targeted therapy for AD.
Search Strategy and Selection Criteria
We searched Google Scholar, PubMed, the Chinese National Knowledge Infrastructure database, the Chinese Medical Journal full-text database (yiigle.com), and the Excerpta Medica (EMBASE) database, using the following keywords: “Alzheimer’s disease,” “AD,” “ferroptosis,” “regulated cell death,”and “Fe2+” from 2013 to 2023.The literature search focused on articles published between 2013 and 2023 and was performed by the author LF on March 1, 2023.The following categories of publications will not be included in this narrative review: 1) Literature that lacks experimental data on the relationship between ferroptosis and AD; 2) publications that have been previously published or for which the full text is unavailable; and 3)conference papers, case reports, patents, and similar types of publications.
Ferroptosis
The definition of ferroptosis
Ferroptosis is a unique form of iron-dependent RCD that distinguishes itself from other forms of RCD, including apoptosis, necroptosis,pyroptosis, autophagy, and cuprotosis.Researchers have established the main mechanisms underlying ferroptosis.In the presence of Fe2+and/or lipoxygenases (LOXs), the polyunsaturated fatty acid chains (PUFAs) of a cell membrane catalyze LPO and ultimately induce ferroptosis in cells.In addition,GPX4 is an important target that can trigger ferroptosis and represents the core enzyme in the antioxidant GSH system (Dixon et al., 2012).
The discovery of ferroptosis
Dolma et al.(2003) discovered that erastin could induce a new form of cell death in KRAS-mutated cells that did not involve the formation of apoptotic bodies, DNA fragmentation, activation of the cysteinyl aspartate specific proteinase (Caspase) family, or changes in nuclear morphology.This observation led to the inference that erastin-induced cell death represented a novel form of death.Subsequently, researchers found that the application of iron-chelating agents could inhibit this new form of cell death, accompanied by increased levels of ROS (Yagoda et al., 2007; Yang and Stockwell, 2008).Hence, this new form of cell death was officially called ferroptosis (Dixon et al., 2012).In 2018,ferroptosis was included as a new type of RCD (Galluzzi et al., 2018).
Morphological characteristics of ferroptosis
A preliminary investigation demonstrated that ferroptosis is morphologically,biochemically, and genetically distinguishable from apoptosis, necrosis,and autophagy (Dixon et al., 2012).In addition, ultrastructural analysis demonstrated that during ferroptosis, cells commonly undergo morphological changes that are similar to necrosis (Vanden Berghe et al., 2014), including a loss of plasma membrane integrity, cytoplasmic swelling, the swelling of cytoplasmic organelles, and moderate chromatin condensation (Tang et al.,2021; Jhelum and David, 2022; Li and Jia, 2023).In addition, the lipid bilayer undergoes changes in structure, permeability, and fluidity (Li et al., 2020a;Tang et al., 2021).Simultaneously, the mitochondria undergo atrophy, with a reduction or disappearance of the mitochondrial crest, an increase in membrane density, and rupture of the outer membrane (Yagoda et al., 2007;Dixon et al., 2012; Friedmann Angeli and Conrad, 2018; Lei et al., 2019).The main characteristics of apoptosis, necroptosis, autophagy, pyroptosis, and ferroptosis are given in Table 1.
The mechanism of ferroptosis
Ferroptosis is primarily caused by an imbalance between the generation and degradation of lipid ROS in cells.The accumulation of lipid ROS due to a reduction in the antioxidant capacity of cells may lead to oxidative cell death in a process known as ferroptosis (Chen et al., 2022).This process can be induced by both exogenous and endogenous pathways.Exogenous pathways include the inhibition of System Xc–(cystine/glutamate transporter) on the cell membrane or activation of the transferrin and transferrin receptor 1.The endogenous pathway involves blocking the activation of the intracellular antioxidant enzyme GPX4 (Li et al., 2020a; Tang et al., 2021).
Inhibition of cystine/glutamate transporters
The cystine/glutamate reverse transporter (System Xc–) is a dimer and transmembrane amino acid transporter composed of the light chain subunit Slc3a2 and the heavy chain subunit Slc7a11.Research has shown that Slc7a11(solute carrier family 7 member 11, also known as xCT) is the main functional subunit and is located in 4q28–q32 with 14 exons (Bassi et al., 2001; Koppula et al., 2018; Lin et al., 2020; He et al., 2022).The protein is formed from 501 amino acids, including 12 transmembrane domains, with its N- and C-termini located in the cytoplasm (Bassi et al., 2001; Koppula et al., 2018; Lin et al.,2020).SLC7A11 plays a crucial role in the regulatory network of ferroptosis,serving as one of the most important regulatory factors upstream.As a reverse transporter, SLC7A11 plays a key mechanistic role in the occurrence of ferroptosis (Yan et al., 2022).Slc7a11 is a protein responsible for transporting amino acids across the plasma membrane (Bassi et al., 2001; Lin et al., 2020)and facilitates the exchange of intracellular glutamic acid with extracellular cystine in a 1:1 ratio (Lin et al., 2016).Following intracellular transport,cysteine undergoes reduction to L(+)-cysteine.This amino acid is rate-limiting and essential for the production of GSH, an important cytoprotective agent(Koppula et al., 2021).L(+)-cysteine is catalyzed by adenosine triphosphatedependent glutamate cysteine ligase to form γ-glutamyl cysteine, which is then combined with glycine and catalyzed by glutathione synthase to synthesize GSH (Yang et al., 2014).
GSH exists in two forms: a reduced form (GSH) and an oxidized form(glutathione disulfide: GSSG).GSSG is formed from two GSH molecules by sulfhydryl dehydrogenation.GSH is the primary source of sulfhydryl in most living cells and is a crucial antioxidant in animal cells.Simultaneously, GSSG serves as a necessary cofactor for glutathione peroxidase (GPX) (Ekoue et al., 2017) and plays a key role in reducing H2O2to H2O, thus maintaining the balance of intracellular ROS (Tang et al., 2021).GPX4 is a core member of the GPX family and converts L-OOH into L-OH, inhibits the generation of lipid ROS,and helps to resist ferroptosis in cells (Flohé et al., 2011).Oxygen-free radicals and LPO are essential for metabolism in the body and maintain a balance that supports various physiological and biochemical reactions, including immune responses (Tang et al., 2021).However, a disruption in this balance can lead to metabolic disorders and immune dysfunction.This disruption triggers a cascade of oxygen-free radicals, resulting in the breakdown of biofilms and their functions, ultimately causing cellular deterioration and damage to the body.This process is known as LPO (Tang et al., 2021), a process that occurs when ROS oxidizes biofilms.Erastin is a well-known inducer of ferroptosis that works by inhibiting the transporter activity of system Xc–.Erastin inhibits the import of cystine through SLC7A11, thus resulting in a reduction in the intracellular levels of GSH.Reduced levels of GSH cause an increase of LPO and ultimately ferroptosis (Iida et al., 2021; Koppula et al., 2021).The Xc–-GSH-GPX4 signaling axis serves as the primary defense mechanism in mammalian cells against the occurrence of ferroptosis.This inhibition leads to a breakdown of the cellular defense system against ferroptosis, thus resulting in the excessive accumulation of lipid peroxides on the cell membrane and ultimately inducing ferroptosis in cells (Dixon et al., 2012; Chen et al., 2020b;Xu et al., 2020; Jiang et al., 2021b).Upon activation, tumor protein 53 (p53)can bind to the promoter region of Slc7a11, thus leading to the inhibition of its transcriptional activity.This reduces the antioxidant capacity of cells and increases sensitivity to ferroptosis (Guan et al., 2020; Luo et al., 2021).
Imbalance in iron homeostasis
The impairment of iron metabolism is considered a crucial factor in ferroptosis(Zille et al., 2017).In normal circumstances, extracellular Fe3+combines with transferrin to create a complex that binds to transferrin receptor 1 on the plasma membrane.This process results in the formation of vesicles that can enter cells via endocytosis (Chen et al., 2021c).Fe3+dissociates from transferrin in the highly acidic nuclear endosomal environment, while the six-transmembrane epithelial antigen of prostate 3 reduces Fe3+to Fe2+.Subsequently, Fe2+is transported to the mitochondria via mitochondrial ferritin transport and participates in vital biological processes including the tricarboxylic acid cycle and the mitochondrial respiratory chain.A portion of Fe2+is transported to the cytoplasmic labile iron pool (LIP) by divalent metalion transporter-1 or ZIP8/14; these are members of the ZRT/IRT-like protein(ZIP) family.Excess ferritin is stored in the form of ferritin (Zheng and Conrad,2020).Nuclear receptor coactivator 4 is a specific protein that facilitates the transportation of ferritin to the lysosomes for autophagic degradation, thus leading to the release of Fe2+.This process is commonly referred to as “iron autophagy” and is responsible for the release and recovery of iron (Goodall and Thorburn, 2014; Gao et al., 2016; Hou et al., 2016; Zhou et al., 2020).Fe2+, especially in LIP, is highly reactive and can produce harmful radicals,including hydroxyl radicals (OH·) and hydroperoxy radicals (O2·) via the Fenton reaction with H2O2.These radicals can activate arachidonate LOX and react with PUFAs in cell membranes and plasma membranes, thus leading to the production of a large amount of lipid ROS and ultimately resulting in cell death (Mancias et al., 2014; Chen et al., 2022).The enzyme LOX, especially 15-LOX, has been identified as a crucial factor in LPO and ferroptosis (Seiler et al., 2008; Yang et al., 2016; Kagan et al., 2017).LOX plays a vital role in the metabolism of unsaturated fatty acids by introducing molecular oxygen to arachidonic acid (AA), oxygenating linoleic acid and linolenic acid, and ultimately producing peroxide (Wang et al., 2021a).Lipidomics research has demonstrated that oxidized phosphatidyl ethanolamine (PE), which contains AA, is a significant phospholipid that can trigger ferroptosis in cells (Yang et al., 2014; Doll et al., 2017; Kagan et al., 2017).A previous study showed that the position of the double bond, and with the saturation level and chain length of fatty acids, are also essential for the induction of ferroptosis (Doll et al., 2017).In addition, research has shown that macrophages and M1-type microglia expressing inducible nitric oxide synthase are highly resistant to ferroptosis, and that the protective mechanism involved is related to 15-LOX(Kapralov et al., 2020).
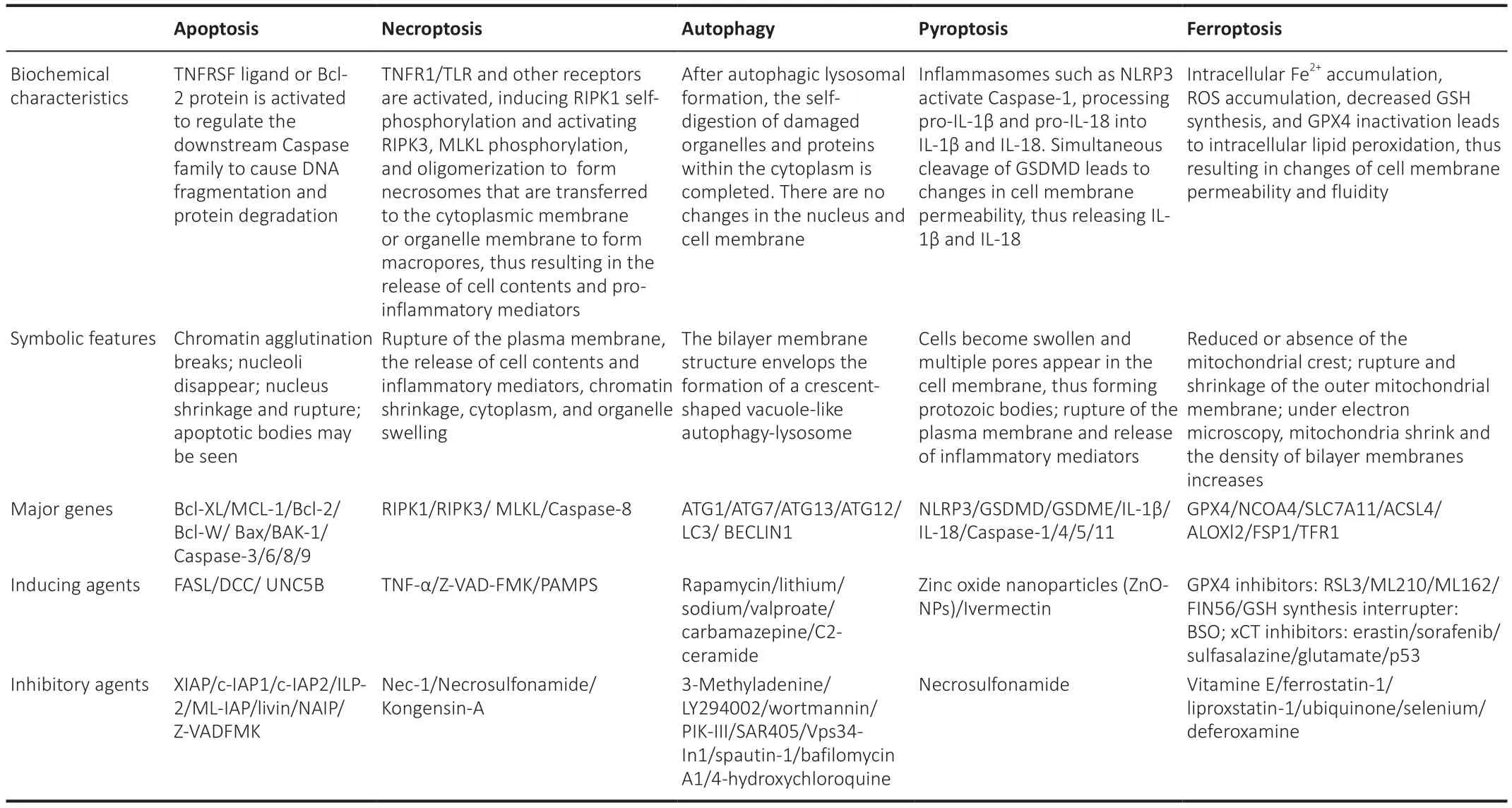
Table 1 |Comparison of RCD characteristics on apoptosis, necroptosis, autophagy, pyroptosis and ferroptosis
In a previous study, Skouta et al.(2014) found that the second-generation ferrostatin compound SRS11-92 was 15-fold more effective than ferrostatin-1 in terms of inhibiting LOX activity and slowing down the accumulation of phospholipid hydroperoxides (PLOOHs; Cotticelli et al., 2019).PUFA is the substrate necessary for the synthesis of LPO but needs to be esterified to membrane phospholipids and oxidized before it can induce ferroptosis(Kagan et al., 2017).During ferroptosis, LPO mainly affects esterified PUFAs and not free PUFAs (Kagan et al., 2017).In the presence of Fe2+, PLOOH can produce harmful lipid-free radicals, especially the alkoxyl group (RO–).Lipidfree radicals can transfer protons adjacent to PUFAs and re-initiate LPO.This process can ultimately result in the oxidation, shortening, or even breakage of carbon chains.Finally, PLOOH can be decomposed into malondialdehyde and 4-hydroxynonenal (4-HNE; Hassannia et al., 2019).These by-products can damage the lipid bilayer of cells, thus reducing the fluidity of membrane lipids and ultimately resulting in cell death (Chen et al., 2021c).4-HNE, also known as the “second messenger for free radicals,” serves as the primary bioactive marker for LPO.Moreover, malondialdehyde and 4-HNE have the ability to covalently bind to basic amino acids [such as lysine, histidine, arginine, and L(+)-cysteine] in cells, ultimately altering their structure and function (Shah et al., 2018).
Blocking the activation of GPX4
Cell death can occur in various forms, including apoptosis, necrosis,pyroptosis, and ferroptosis.Of these forms, ferroptosis is specifically characterized by iron-dependent LPO accumulation.The regulation of LPO production and clearance is considered the main determinant of ferroptosis(Hassannia et al., 2019).In organisms, the antioxidant system plays a key roles in defending and repairing oxidative damage.While antioxidant enzymes,such as superoxide dismutase, catalase, and GPX, are capable of removing excessive free radicals and peroxides from the cytoplasm, these enzymes cannot directly reduce peroxides in biofilms.Therefore, these enzymes are unable to protect biofilms from peroxidation damage.In a previous study,Ursini et al.(1982) isolated and purified a protein known as “peroxidation inhibitor protein” from various organs in mice and pigs.This protein, known as GPX4 or phospholipid hydroperoxide GSH peroxidase (PHGPx), directly protects phospholipids on biofilms from peroxidation damage and can specifically reduce PLOOH on the membrane when facilitated by the reducing substrate GSH (Brigelius-Flohé, 2006).
GPX4 is a water-soluble protein with a molecular weight of approximately 19 kDa and encoded by a located at 19p13.3 in the human genome (Kelner and Montoya, 1998).GPX4 is the fourth member of the GPX family, is encoded by seven exons, and composed of 170 amino acid residues (Sunde and Hadley, 2010).The active center of GPX4 contains selenocysteine (Sec)residues, and functions as a selenoenzyme (Knopp et al., 1999).GPX family members, including GPX1-GPX8, have been identified in mammals (Passaia et al., 2014).However, only GPX4 has demonstrated the ability to scavenge PLOOH.Numerous studies have shown that GPX4 serves as a core regulator of ferroptosis and is considered a key indicator of cellular ferroptosis (Anandhan et al., 2020).In fact, GPX4 is often referred to as the “star molecule” in ferroptosis research.Although ferroptosis was first discovered in 2012 (Dixon et al., 2012), the specific role of GPX4 has yet to be fully elucidated.In 2014,researchers discovered that the depletion of GSH resulted in the deactivation of GPXs, as revealed by targeted metabolomics analysis (Yang et al., 2014).In addition, these authors used chemical proteomic analysis to identify GPX4 as a crucial molecule (Yang et al., 2014).The knockdown or overexpression of GPX4 by 12 types of ferroptosis-targeting drugs suppressed ferroptosis,but did not affect the other types of cell death targeted by 11 different therapeutic agents (Yang et al., 2014).These findings suggested that GPX4 is a crucial regulator of ferroptosis (Yang et al., 2014).While various signaling pathways can trigger ferroptosis, they all ultimately impact the activity of GPX4, leading to a reduction in cellular antioxidant capacity and an increase in LPO, ultimately resulting in ferroptosis (Flohé et al., 2011; Yang et al., 2014).GPX4 catalyzes the reduction of PLOOH to PLOH by using GSH as a reducing substrate.There are two pathways for removing PLOOH from biofilms.The first pathway is the cytosolic glutathione peroxidase pathway, in which phospholipase A2 is activated by peroxide and Ca2+, thus leading to cleavage of the peroxide-acid chain.The cleaved PLOOH is then reduced to PLOH by either cytosolic glutathione peroxidase or phospholipid hydroperoxide GPX, both members of the GPX family.The second pathway is the GPX4 pathway; this involves the direct reduction of PLOOH to PLOH by GPX4 and the cleavage of fatty acid chains by phospholipase A2.This pathway is crucial for protecting the integrity of biofilms, as it converts activated PLOOH to deactivated phosphatidylcholine, a major component of cellular membranes.This interference with free radical chain reactions and the inhibition of LPO processes reduces the occurrence of ferroptosis, thus demonstrating the importance of the GPX4 pathway (Flohé et al., 2011; Yang et al., 2014; Tang et al., 2021).
During catalysis, GSH provides hydrogen, which is subsequently oxidized into GSSG.In vivo, GSSG can be reduced to GSH for recycling by GSH reductase(with the hydrogen provided by NADPH) (Hassannia et al., 2019; Chen et al., 2021a).The catalytic reaction rate of GPX4 is positively correlated with enzyme concentration, GSH levels, and peroxide levels.Notably, GPX4 cannot be saturated by its substrate GSH.The inhibition of LOX attenuated RSL3-induced ferroptosis, while the inhibition of cyclooxygenase had no specific effect (Kagan et al., 2017).RSL3 is a small molecule compound that inhibits GPX4 (Yang et al., 2014) and represents the target protein for RSL3 (Chen et al., 2015).RSL3 contains the electrophilic compound 2-chloroacetamide,which reacts covalently with Sec, a nucleophilic active site in GPX4.This interaction leads to the irreversible inactivation of the GPX4 enzyme (Yang et al., 2016) and causes a shift in the redox balance of cells toward ROS accumulation, oxidative stress, and ultimately ferroptosis (Shintoku et al.,2017).Knocking down the Gpx4 gene increased sensitivity to RSL3-induced ferroptosis, while the overexpression of GPX4 increased cell resistance to ferroptosis (Imai et al., 2017).Further research found that RSL3 treatment induced ferroptosis in a manner that was similar to GPX4 inactivation, thus providing further evidence that RSL3 suppresses GPX4-induced ferroptosis(Yang et al., 2014).
The epoxy chloropropane Kelch sample-related protein-1 (Keap1)-nuclear factor erythroid-derived 2-like 2 (Nrf2)-antioxidant response element (ARE)signaling pathway is a crucial endogenous defense mechanism that plays a vital role in protecting cells against oxidative stress caused by external or internal factors.This pathway is initiated by Nrf2, which activates ARE and regulates antioxidant reactions (Ma, 2013).Under normal circumstances, Nrf2 binds primarily to Keap1.However, under conditions of oxidative stress, Nrf2 separates from Keap1 and enters the nucleus to initiate the transcription of antioxidant genes (Kajarabille and Latunde-Dada, 2019; Chen et al., 2020a).During transcription, Nrf2 only mediates theGPX4gene (Doll et al., 2017).When stimulated by ROS or induced electrophiles, Nrf2 can activate the expression of downstream II phase detoxification enzymes, including GSHPx, superoxide dismutase, quinone oxidoreductase 1, and NQO1 (Na and Surh, 2008), and a variety of antioxidant stress proteins such as GPX4 (Zhang et al., 2018b).Nrf2 induces the expression of xCT and GPX4 in the GSH antioxidant system, thus helping to prevent ferroptosis (Chen et al., 2020a).Heat-shock-protein family A member 5 (HSPA5) is a molecular chaperone protein that is predominantly expressed on the endoplasmic reticulum (ER).Under conditions of ER stress, HSPA5 promotes cell survival.A previous study showed that HSPA5 could interact with GPX4 to prevent its degradation by inhibiting ubiquitination, thereby prolonging the half-life of cells (Zhu et al.,2017).Unfortunately, it remains unknown as to which E3 ubiquitin ligase binds to the competing binding sites of GPX4 and HSPA5.However, evidence clearly indicates that GPX4 plays key roles in ferroptosis.Figure 1 illustrates the mechanisms underlying ferroptosis in detail.
Other causes of ferroptosis
Initially, AA and PE were regarded as the primary substrates for LPO (Kagan et al., 2017).Furthermore, lipidomic research demonstrated that PEs and phospholipids play a crucial role in promoting ferroptosis via oxidation (Doll et al., 2017; Kagan et al., 2017).However, a recent study proposed that this process may also be reliant on PUFAs (Doll et al., 2019).The process by which cells acquire PUFAs occurs in two steps: first, essential fatty acids are acquired from the diet; then, these fatty acids are converted via the combined action of desaturase and elongase (Guillou et al., 2010).Previous research indicated that the knockdown of fatty acid desaturase 2 resulted in HUH7 hepatoma cells developing resistance to RSL3-induced LPO (Vriens et al., 2019).In addition, the inhibition of acetyl-coenzyme A (CoA) carboxylase via the use of adenosine 5′-monophosphate-activated protein kinase or acetyl-CoA carboxylase inhibitors significantly enhanced protection against ferroptosis.Research suggested that the production of malonyl-CoA in a manner that is facilitated by acetyl-CoA carboxylase, is essential for the elongation of PUFAs(Shimada et al., 2016; Lee et al., 2020).Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are known to catalyze the LPO of PE that has been converted from epinephrine(Miao et al., 2022).The genetic deletion ofACSL4andLPCAT3genes results in a significant increase in the resistance to ferroptosis (Doll et al., 2017; Kagan et al., 2017).In addition, previous studies demonstrated that ACSL4 plays a role in promoting ferroptosis by upregulating the expression of 5-hydroperoxy eicosatetraenoic acid, which is known to be lipotoxic and serves as a reliable biomarker for ferroptosis (Dixon et al., 2012; Yang et al., 2014).In conclusion,the manipulation of the synthesis or degradation pathways of PUFAs will influence cells’ susceptibility to ferroptosis.
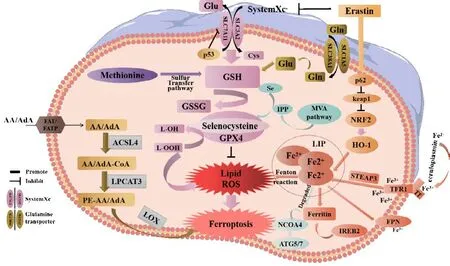
Figure 1 |The mechanisms underlying ferroptosis.
The mevalonate (MVA) pathway may regulate the maturation of Sec tRNA, which in turn affects GPX4 and induce ferroptosis in cells.Hydroxy methylglutaryl CoA reductase inhibitors, which block the rate-limiting enzyme in the MVA pathway, are known to impair the effective translation of GPX4 and increase the likelihood of ferroptosis (Shimada et al., 2016; Viswanathan et al., 2017).Sec is an amino acid found at the central active site of GPX4.The embedding of Sec into GPX4 requires special transporters known as Sec tRNA (Min et al., 2018; Wang et al., 2021b).The maturation of Sec tRNA requires isopentenyl transferases to transfer the isopentene group from isopentenyl pyrophosphate (IPP) to Sec tRNA precursors.IPP is a product of the MVA pathway.Thus, the MVA pathway may affect the synthesis of Sec tRNA by downregulating IPP, which further interferes with the activity of GPX4 to induce ferroptosis (Cardoso et al., 2017).IPP is a crucial precursor to the production of squalene and coenzyme Q10 (CoQ10) and also serves as a substrate for the Sec-tRNA catalysis of isoprenylation reactions, in which it acts as a limiting factor (Moosmann and Behl, 2004; Friedmann Angeli and Conrad, 2018).A recent study demonstrated that squalene exhibits antiferroptosis properties in certain types of cancer cells (Zheng and Conrad,2020).In addition, CoQ10 can act as an endogenous inhibitor of ferroptosis and is metabolized by the MVA pathway.However, the ferroptosis inducer FIN56 may activate squalene synthase, ultimately resulting in the depletion of CoQ10 (Shimada et al., 2016).In a previous study, Bersuker et al.(2019)discovered that CoQ10 has the ability to capture lipid ROS via panthenol,its reduced form.Ferroptosis suppressor protein 1, previously known as apoptosis-inducing factor mitochondria associated 2, was previously shown to be recruited to the plasma membrane following myristoylation.Subsequently,ferroptosis suppressor protein 1 inhibited the release of LPO by catalyzing CoQ10 regeneration via NADPH.Thus, ferroptosis suppressor protein 1/CoQ10 can prevent ferroptosis without relying on the GPX4 pathway (Bersuker et al., 2019; Doll et al., 2019).This was the first report of an enzyme-catalyzed system that compensates for the loss of GPX4 (Bersuker et al., 2019; Doll et al., 2019).
The methionine (Met) pathway is crucial for biosynthesis and metabolismin vivo.Methylation plays an important role in this pathway.Met is an essential amino acid in humans and is the primary source of methyl donation.Met cannot be generated internally; therefore, it must be obtained from external sources (Li et al., 2020b).Met is transformed into S-adenosyl Met via the action of adenosine triphosphate and Met activase.This compound then undergoes methylation via methyltransferase to generate a diverse range of physiologically active substances (Sun et al., 2018).S-adenosyl Met provides methyl groups and is then converted into homocysteine, which accepts methyl groups from 5-methyltetrahydrofolate.Met is regenerated through the action of Met synthetase in a process known as the Met cycle.Met serves as a precursor for the synthesis of GSH.During oxidative stress, met can be converted to cysteine through the sulfur transfer pathway to synthesize GSH(Sun et al., 2018; Homma et al., 2022).This process helps GPX4 to inhibit the production of lipid ROS and prevent oxidative cell damage.Thus, the sulfur transfer pathway can inhibit the occurrence of ferroptosis; the detailed mechanism involved in this process is shown in Figure 2.
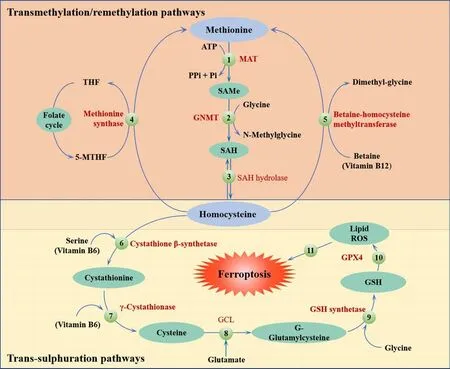
Figure 2 |The sulfur transfer pathway can inhibit the occurrence of ferroptosis.
The heme oxygenase (HO)-1 pathway involves three isoforms of HO in mammals: the oxygen stress-induced type (HO-1), the constitutive type(HO-2), and the as yet unidentified type (HO-3).HO-1, also known as heat shock protein 32, is a stress response protein that is expressed at relatively low levels under physiological conditions (Stocker and Perrella, 2006).However, HO-1 can be induced by various stimuli in multiple tissues and organs and then participate in various pathologies and stress states (Stocker and Perrella, 2006).HO-1 is a crucial factor in sourcing intracellular iron and regulating iron metabolism.Furthermore, HO-1 deficiency can indirectly lead to LPO-induced ferroptosis.The daily iron requirements of humans are met by the decomposition of senescent red blood cells that are engulfed by macrophages.This form of decomposition is primarily facilitated by the catabolism of heme; HO-1 is a rate-limiting enzyme in this process (Pérezde-Puig et al., 2013) and decomposes heme into CO, Fe2+, and bilirubin(Maines, 1997; Wang et al., 2013).Excessive amounts of these by-products can cause HO-1 to shiftfrom a cell protector to a perpetrator that can cause DNA damage, gene mutations, and potentially, cell death (Kwon et al., 2015;Kajarabille and Latunde-Dada, 2019).A previous study, involving myocardial cells from mice, showed that the injection of adriamycin reduced the levels of heme but increased the levels of Fe2+.However, the use of heme oxygenase inhibitors, or knocking down the upstream regulatory molecule NRF2,reduced the accumulation of Fe2+in cardiomyocytes, thereby reducing the occurrence of ferroptosis (Fang et al., 2019).According to these findings,ferroptosis, induced by Fe2+release from the HO-1-induced decomposition of heme, is the primary cause of adriamycin-induced cardiotoxicity in mice (Fang et al., 2019).Thus, the specific role of HO-1 is determined by the cellular levels of iron and ROS.Figure 3 illustrates the mechanisms underlying the effects of NRF2/HO-1 in ferroptosis.
Cuprotosis and ferroptosis
Copper is a crucial trace element for human physiology.However, the excessive intake of copper can damage cells and even cause cells to die.This is because copper can induce intracellular ROS and inhibit proteasomal function,thus triggering cellular apoptosis (Chen et al., 2021b) and cuprotosis (Tsvetkov et al., 2022).According to a recent study, intracellular Cu2+ions are able to act as novel inhibitors of GPX4.Cu2+can reduce the levels of GPX4 protein without affecting GPX4 gene expression during ferroptosis.In addition, Cu2+chelating agents have been found to reverse the erastin-induced reduction of GPX4 protein (Xue et al., 2023).Xue et al.(2023) reported that Cu2+promotes ferroptosis by facilitating the binding of cysteine residues C107 and C148 in GPX4 to autophagy receptor TAX1BP1.This mechanism is independent of ROS production and iron metabolism but is known to increase the ubiquitination of GPX4 (Xue et al., 2023).These findings indicate that Cu2+may provide novel therapeutic options.For example, the inhibition of Cu2+chelation may enhance ferroptosis in cancer cells or prevent diseases caused by ferroptosis(Xue et al., 2023).In addition, these findings provide preliminary evidence that cuprotosis and ferroptosis are closely related.
Ferroptosis and Alzheimer’s Disease
The association of ferroptosis and Alzheimer’s disease
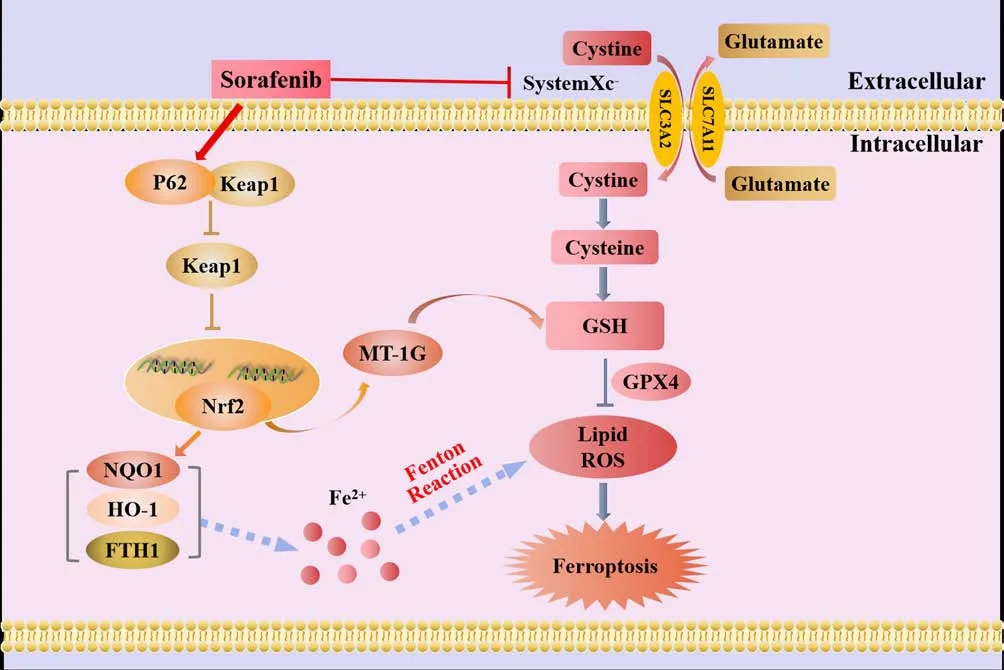
Figure 3 |The mechanistic effects of Nrf2/HO-1 in ferroptosis.
Numerous studies have shown that abnormal iron metabolism can be associated with the development of AD (Kwiatek-Majkusiak et al., 2015; Bulk et al., 2018; Lee and Lee, 2019; Yan and Zhang, 2019; Villalón-García et al.,2023).The brain tissues of both AD patients and AD model mice have been reported to show features related to ferroptosis, including abnormal iron metabolism, glutamate excitotoxicity, and the accumulation of lipid ROS (Lei et al., 2019).Research has also shown that AD patients have higher levels of iron in the hippocampus, cortical lobe, and basal ganglia than control subjects.The amount of iron and ferritin in brain tissue has also been associated with the extent of amyloid deposition (Kwiatek-Majkusiak et al., 2015; Bulk et al., 2018; Lee and Lee, 2019).In addition, AD patients also show elevated concentrations of malondialdehyde and 4-HNE in various brain regions, along with the downregulated expression of GPX4 (Dare et al., 2020).A previous study demonstrated that the intramuscular injection of deferoxamine, an iron-chelating agent, helped to improve the daily living abilities of patients with AD (Rogers and Lahiri, 2004).Glutamate excitotoxicity is a known factor in the development of AD.The dysfunction of system Xc–during ferroptosis is known to cause an increase in extracellular glutamate concentration that can lead to AD (Zhang et al., 2020).Furthermore, Li et al.(2018) reported that the expression of ferroptosis-related proteins (GPX4, SLC7A11, ACSL4,phosphatidylethanolamine binding protein 1) was higher in the hippocampi of a mouse model of AD model in which PSEN1 had been knocked down than in healthy mice, thus suggesting that ferroptosis is closely related with neurodegenerative diseases such as AD.Consequently, it follows that ferroptosis may be an important mechanism in the development of AD.
Imbalance in iron homeostasis and AD
An imbalance in iron homeostasis is known to contribute to the development of AD, and an excess of iron can aggravate the oxidative damage and cognitive dysfunction associated with disease.Previous studies have found that patients with a standardized diagnosis of AD have elevated levels of iron in their inferior temporal cortex.While iron levels are not strongly correlated with the extent of neuropathological changes in AD, they are closely linked to the rate of cognitive decline (Ayton et al., 2020).In addition, a previous study reported that the expression of ferroportin (Fpn) is significantly downregulated in the brains of APPswe/PSEN1dE9 (APP/PS1) mice and individuals with AD.The restoration of Fpn has been shown to ameliorate ferroptosis and memory impairment in APP/PS1 mice (Bao et al., 2021).Currently, Fpn is the only known protein that can facilitate the excretion of iron from cells (Jiang et al., 2021a).The hybridization of Fpnfl/fland NEX-Cre mice led to the genetic deletion of Fpn in neurons of the hippocampus and cortex of Fpnfl/fl/NEXCre mice, thus resulting in atrophy and memory dysfunction.In addition,the typical morphological and key biochemical characteristics of ferroptosis were observed in these animals.The Administration of ferroptosis-specific inhibitors has been shown to effectively reduce the neuronal death and neurological dysfunction caused by Aβ aggregation (Bao et al., 2021).In addition, enrichment analysis of a ferroptosis-related RNA-sequencing dataset revealed high enrichment of differentially expressed genes related to AD (Bao et al., 2021).
The majority of APP undergoes cleavage via the non-amyloidogenic pathway,initially by α-secretase and followed by γ-secretase.However, the cleavage of APP by β-secretase and then γ-secretase can result in the production of Aβ (Altamura and Muckenthaler, 2009; Ward et al., 2014).Cells possess a protease with shearing activity that activates specific sites in non-biologically active precursor proteins during the production and secretion of many proteins; this can result in the acquisition of specific biological functionality.Furin is a crucial proprotein convertase in mammals that is responsible for the proteolytic maturation of various prohormones and proproteins within the secretory pathway (Zhang et al., 2022).Furin has been implicated in the pathogenesis of AD via various mechanisms.For example, furin enhances the activity of α-secretase which can reduce the production of Aβ in APP metabolism (Rowan et al., 2003; Nykjaer et al., 2004).In addition, furin can mediate the cleavage of propeptides during the processing of β-secretase propeptides, thus inhibiting the synthesis of β-secretase and reducing the formation of Aβ, ultimately inhibiting plaque deposition (Guillemot et al.,2013).Furin can also catalyze the transformation and maturation of nerve growth factor and brain-derived neurotrophic factor which are known to exert neuroprotective properties (Teng et al., 2005; Lu et al., 2013).Research has also shown that furin can regulate the activity of low-density lipoprotein reception-related protein, a protein known to influence the metabolism of apolipoprotein E (ApoE) (Kirkland et al., 2007).In addition, furin is known to influence ER stress, thereby regulating apoptosis and autophagy.According to previous studies (Ward et al., 2014; Zhang et al., 2022), high levels of iron in the body can reduce the activity of furin.This arises because the transcription of furin is known to be affected by the levels of iron in cells, thus leading to lower concentrations of furin protein (Ward et al., 2014).These lower concentrations of furin can increase the activity of β-secretase and enhance the amyloidogenic pathway; this process is known to be associated with the progression of AD (Ward et al., 2014).These findings suggest that targeting this imbalance may represent a promising treatment approach for disease(Bao et al., 2021).A visual representation of the mechanism underlying thus imbalance and its relationship with AD is shown in Figure 4.

Figure 4 |The relationship between iron homeostasis imbalance and AD.
The association of LPO with AD
AD is characterized by several pathological features, including the deposition of Aβ outside cells, the formation of NFTs inside cells due to the hyperphosphorylation of tau protein, the loss of synapses, mitochondrial dysfunction, oxidative stress, metabolic disorders, neuroinflammation,and the loss of cholinergic neurons (Ryu et al., 2019; Webers et al., 2020).Research has indicated a possible connection between Aβ deposition and LPO.A previous study involving a rat model of AD induced by Aβ25–35revealed damage caused by oxidative stress, LPO, and changes in acetylcholine activity in the hippocampus (Dare et al., 2020).The deposition of Aβ and the hyperphosphorylation of tau are both known to initiate ER stress (Gerakis and Hetz, 2018), which can further lead to the hyperphosphorylation of Tau protein, thus exacerbating AD and generating a harmful cycle (Ho et al.,2012).In addition, ER stress and the excessive buildup of ROS in the brain can promote LPO, thus resulting in the formation of 4-HNE; this can hinder the dephosphorylation of tau protein (Zhou et al., 2013).Butterfield and Boyd-Kimball discovered that AD patients had elevated levels of oxidative stress in regions of the brain that were rich in Aβ1–42and that the levels of 4-HNE in the brain region that was rich in Aβ-polypeptides were significantly increased(Butterfield and Boyd-Kimball, 2018).These findings provide preliminary evidence that Aβ-polypeptide leads to LPO in the AD brain.When compared with a control group, mice with tamoxifen-induced GPX4 deficiency (known as the Gpx4BIKO mouse model) exhibited notable impairments in spatial learning and memory function, as well as neurodegenerative changes in the hippocampus (Hambright et al., 2017).In addition, these mice had elevated markers associated with ferroptosis (Hambright et al., 2017).Hippocampal neurodegeneration and neurological dysfunction were shown to be aggravated in Gpx4BIKO mice fed a diet that was deficient in vitamin E, a fat-soluble antioxidant that acts against ferroptosis.However, treatment with liproxstatin 1, a small-molecule ferroptosis inhibitor, was shown to significantly improve these symptoms (Hambright et al., 2017).These results suggest that ferroptosis does occur in the brains of animal models of AD and may contribute to LPO via endogenous pathways (Dare et al., 2020).
LPO and Aβ deposition
In the early stages of AD, damage to the mitochondria leads to an accumulation of intracellular ROS, which in turn causes mitochondrial dysfunction (Di Meo et al., 2016) and promotes the deposition of Aβ(Swerdlow et al., 2014).A previous study showed that Aβ1–42induced elevated concentrations of intracellular Ca2+and ROS in U251 cells (Wang et al.,2020).Furthermore, Aβ1–42was shown to alter the morphology of astrocytes,activate protein kinase R-like ER kinase/eukaryotic initiation factor 2α (PERK/ELF2α)-dependent ER stress, and regulate the release of interleukin-1β and tumor necrosis factor, thus leading to neuroinflammation and apoptosis and ultimately exacerbating AD (Hong et al., 2016).Han et al.(2021) discovered that PS1 mutation leads to an increase in ER-mitochondrial interactions, the overproduction of ROS, and a reduction in mitochondrial membrane potential.These findings suggested that PS1 may play a crucial role in regulating and maintaining ER-mitochondrial interactions via the atlantique 2 protein, a protein known to be significantly upregulated in the brains of both 5×FAD mice and AD patients (Han et al., 2021).The induction of ER stress leads to Aβ deposition and the hyperphosphorylation of tau protein (Ho et al., 2012; Xu et al., 2019), as well as an increase in ROS levels and the activation of LOXs.This process ultimately results in ferroptosis in cells via the peroxidation of PUFA (Kapralov et al., 2020).Nicotinamide adenine dinucleotide phosphate oxidase (NOX) is the only enzyme that specifically generates ROS (Magnani and Mattevi, 2019).High levels of ROS derived from NOX hyperactivity can lead to genetic instability, excessive proliferation, activation of the DNA damage response, proliferative senescence, and apoptosis (Crosas-Molist et al., 2017; Ju et al., 2017).NOX is an enzyme complex composed of various subunit enzymes.Among these enzymes, NOX4 stands out as the only member of its protein family with constitutive activity (Nisimoto et al., 2010).It plays a critical role as a significant source of ROS in the central nervous system (Drummond et al., 2011).This activity is catalyzed by oxidative stress and contributes to a range of pathophysiological processes (Drummond et al.,2011).Previous research showed that the expression levels of NOX4 protein are substantially elevated in the astrocytes of both AD model mice and AD patients with cerebral cortex damage; this resulted in increased metabolic damage, the promotion of ROS production, and the induction of ferroptosis(Reis et al., 2020; Park et al., 2021).One of the main byproducts of LPO is 4-HNE, a highly reactive electrophilic aldehyde (Shoeb et al., 2014).This aldehyde has the ability to modify the Aβ peptide by inducing misfolding and aggregation (Bieschke et al., 2006; Liu et al., 2008; Butterfield et al., 2010).Studies have detected the presence of 4-HNE in the brains of AD patients at different stages of the disease (Bradley et al., 2010; Ates et al., 2020), thus indicating a potential interaction between Aβ and ROS, particularly LPO.Figure 5 illustrates the mechanisms underlying LPO and the deposition of Aβ.
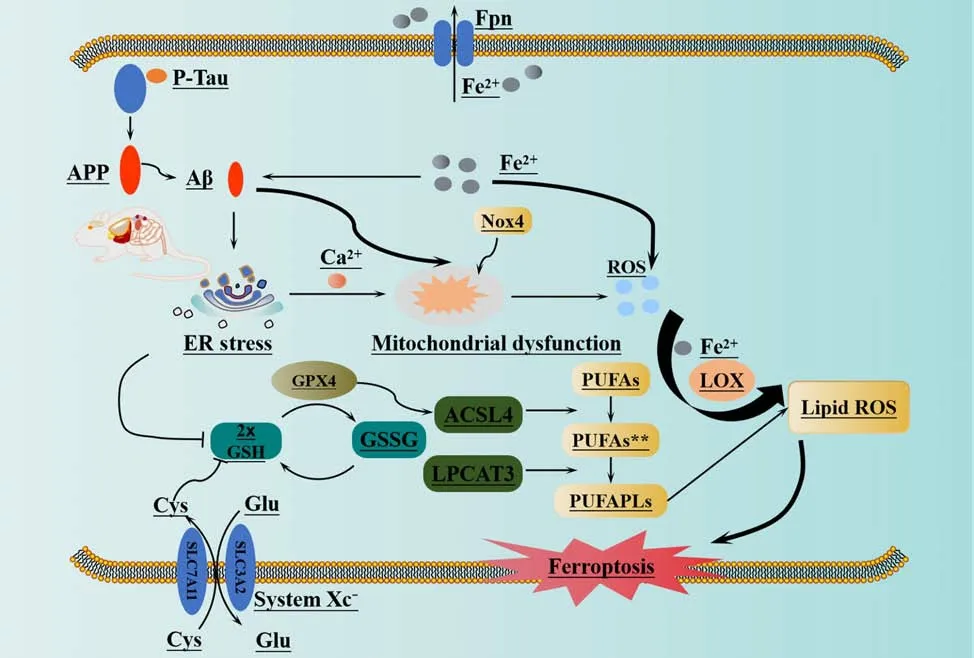
Figure 5 |Ferroptosis and beta amyloid deposition in Alzheimer’s disease.
LPO and the hyperphosphorylation of tau
NFTs are a defining characteristic of tauopathies such as AD (Rao and Adlard,2018).These tangles are primarily composed of paired helical filaments made up of hyperphosphorylated tau protein.The tau protein is an intrinsically unfolded and highly soluble phosphoprotein that is encoded by theMAPTgene (Spillantini and Goedert, 2013).Dysregulation in cerebral iron content is reported to be associated with the progression of tau-mediated neurodegeneration.An increase in iron concentration is also correlated with the formation of NFTs in tauopathies (Duce et al., 2010).Furthermore, there is growing evidence for an association between the accumulation of iron in the brain and abnormal tau pathology in AD (Rao and Adlard, 2018).One of the crucial steps in NFT formation is tau hyperphosphorylation; this is thought to arise because of an imbalance between kinase and phosphatase activity.Kinase activity was previously shown to be strongly associated with early tau deposits and tangles in brain samples from AD patients (Rao and Adlard, 2018).Research has also indicated that iron can exert influence on tau kinases, including glycogen synthase kinase 3β (Ferrer et al., 2002),cyclin-dependent kinase-5 (Pei et al., 1998), and mitogen-activated protein kinase (MAPK) (Perry et al., 1999; Ferrer et al., 2001), and may also regulate the activity of phosphatase 2A.In addition, iron may interact with the tau protein via a putative iron binding motif (Egaña et al., 2003).The inhibition of c-Jun N-terminal kinase and p38 has been shown to prevent erastininduced ferroptosis in HL-60 cells, suggesting a potential link between activation of the MAPK signaling pathway and the promotion of ferroptosis(Yagoda et al., 2007; Yu et al., 2015).Deprivation of cystine in human mammary epithelial (HME) cells was shown to result in increased ferroptosis in cells that express an activated epidermal growth factor receptor mutant.However, blocking the epidermal growth factor receptor or MAPK signaling was shown to protect cells from ferroptosis.These findings indicate that the MAPK signaling pathway may play a role in promoting ferroptosis in cells (Poursaitidis et al., 2017).Studies in cellular and animal models of AD have also demonstrated that MAPK signaling plays a critical role in various pathological events associated with AD, such as tau hyperphosphorylation(Zhu et al., 2000; Kelleher et al., 2007; Li et al., 2012; Giraldo et al., 2014),neuronal damage (Ghasemi et al., 2014; Suwanna et al., 2014; Chen et al.,2016; Cui et al., 2016; Fang et al., 2017; Pierucci et al., 2017; Xie et al., 2017),neuroinflammation (Kim et al., 2013, 2014; Park et al., 2013; Gan et al., 2015;Yang et al., 2015), and synaptic plasticity (Hamilton et al., 2014; Lee and Kim,2017).Consequently, inhibiting the MAPK signaling pathway is considered a promising strategy for the treatment of AD (Lee and Kim, 2017).Iron exists in two oxidation states: Fe2+(redox-active) and Fe3+(redox-inert state) (Rao and Adlard, 2018).Recent electrochemical studies using X-ray photoelectron spectroscopy have suggested that Fe2+may cause reversible conformational changes in tau and promote its aggregation via threonine residues (Ahmadi et al., 2017).Iron dysregulation is hypothesized to be a source of oxidative stress in tauopathies.The interaction between NFTs and iron is reported to act as a source of ROS in neurons (Foroutan et al., 2013).Hyperphosphorylated tau protein promotes LPO; this occurs because phosphorylated tau proteins interact with membrane phospholipids on cell membranes, thus resulting in the formation of a cytotoxic tau-phospholipid complex (Bok et al., 2021).This interaction is believed to be a crucial factor in the induction of tau protein aggregation in cells and can lead to the formation of tau oligomers.These oligomers can then cause the destruction of membrane structure,ultimately resulting in the onset of degenerative diseases (Kawarabayashi et al., 2004; Yao et al., 2022).A previous study demonstrated that AA, an omega-6 polyunsaturated fatty acid, can effectively induce tau polymerization under near-physiological conditions (King et al., 2000).Additionally, lipidomics research has revealed that PE containing AA is a crucial phospholipid that can lead to ferroptosis in cells after oxidation (Yang et al., 2014; Doll et al., 2017;Kagan et al., 2017).
Previous research revealed that P301S tau transgenic mice exhibited significantly increased levels of tau-induced iron overload, LPO, and inflammation; these factors are all associated with ferroptosis (Zhang et al., 2018a).However, treatment with DL-thioctic acid was found to reduce intracellular iron overload and inhibit cell death caused by iron dependence in the cerebral cortex and hippocampus of a mouse model of AD.This finding is consistent with the preventive effect of lipophilic antioxidants on ferroptosis when compared with placebo treatment (Speer et al., 2013; Ma et al., 2016;Basuli et al., 2017; Hambright et al., 2017).In a previous study, Wang et al.(2022) demonstrated that hyperphosphorylated tau protein was associated with ferroptosis LPO, although further research is required to establish a conclusive relationship.
Neuroglial cell iron overload and ferroptosis in AD
Microglia, a type of brain cell, have the ability to store high levels of iron and tend to accumulate iron during disease conditions (Andersen et al.,2014; Song et al., 2018; Absinta et al., 2021).However, the specific effect of iron overload on microglial function, and whether iron-loaded microglia contribute to neurodegeneration, have yet to be elucidated.Ryan et al.(2023)developed a tri-culture system derived from human induced pluripotent stem cells, consisting of microglia, neurons, and astrocytes.These authors discovered that microglia exhibited the most substantial transcriptional response to iron dysregulation.Furthermore, the authors reported that the elimination of microglia resulted in a reduction in neuronal LPO and a significant delay in neuronal death during ferroptotic conditions (Ryan et al.,2023).By performing genome-wide clustered regularly interspaced short palindromic repeats (CRISPR) screening, the team identified a new ferroptosis susceptibility gene,SEC24B, which acted as a regulator in microglia (Ryan et al., 2023).Further research revealed that an SEC24B knockout myeloid cell line exhibited high levels of resistance to ferroptosis and exhibited a significant reduction in LPO after being exposed to ferroptotic stimuli for 2-4 hours.These findings suggested that microglial iron overload and ferroptosis play a crucial role in neurodegeneration (Ryan et al., 2023).Dang et al.(2022)used cell-cell communication analysis to investigate the cellular heterogeneity of AD by analyzing single-cell RNA sequencing data from AD patients and healthy control individuals obtained from the Gene Expression Omnibus(GEO) database (www.ncbi.nlm.nih.gov/geo/).These authors validated the expression patterns and biological functions of differentially expressed genes by considering RNA sequencing data obtained from single-cell RNA sequencing dataset (GSE138852).Furthermore, these authors identifiedFTH1andSAT1as two genes that exert impact on ferroptosis in astrocytes (Dang et al., 2022).
In addition, some researchers consider that Aβ deposition may represent a normal response to elevated levels of iron and that neuritic plaques are a reflection of such neuroprotection; these considerations are supported by experimental data obtained from rats (Bishop and Robinson, 2003a, b).The injection of iron into the brain, either alone or in combination with Aβ,resulted in more severe damage and neuronal loss than injecting Aβ alone.In addition, the co-injection of iron with human Aβ was less toxic than that co-injection with rat Aβ, and exhibited the same toxicity as injecting iron alone (Bishop and Robinson, 2003a, b).Consequently, these researchers concluded that the accumulation of Aβ might represent a defensive response to high levels of iron (Bishop and Robinson, 2003a, b).Previously, researchers identified iron-containing droplet spheres (p-tau+degeneration droplets) that were selectively encased by Aβ in the human brain; this finding provided evidence for the neuroprotective role of Aβ (Robinson and Bishop, 2002).Previous research indicated that microglial activation and the phagocytosis of Aβ occur only when Aβ protein aggregates and transforms into an insoluble form of Aβ (Streit et al., 2018).According to previous studies (Bishop et al.,2004, 2011), microglia are the most efficient cell in the CNS with regards to the accumulation of iron.Lesions begin to develop following neurofibrillary degeneration and microglial dystrophy (Bishop et al., 2011; Streit et al., 2018).This process is then followed by amyloid deposition, neuroinflammation, and an increase in iron levels.Further research has provided a more accurate interpretation of these findings, suggesting that the dissolution of neurons into droplet spheres results in the extracellular release of neuronal iron.The iron released is then rapidly taken up by microglia expressing ferritin; this is followed by the chelation by Aβ (Streit et al., 2022).Gaining an enhanced understanding of these factors may provide new therapeutic targets for the treatment of AD.
Limitations
This review provides a comprehensive portrayal of recent investigations into the functional role of ferroptosis in AD, rather than presenting quantitative data.The content of this review is based on the consensus of opinion across all authors rather than statistical analysis.While not conducted as part of a systematic review, we reviewed the existing literature on the topic and provide an account of the latest research on the role of ferroptosis in AD.
Conclusion
Although several features have been identified in AD that are known to be associated with ferroptosis, further confirmation of the targets associated with ferroptosis is still needed from animal models of AD.Recent studies discovered that the disruption of blood-brain barrier integrity in AD is independent of Aβ and tau and occurs several years before the symptoms of cognitive dysfunction.This disruption is considered an early biomarker of cognitive dysfunction (Sweeney et al., 2018; Nation et al., 2019; Montagne et al., 2020).ApoE is a known genetic risk factor for sporadic AD (Liu et al., 2021).The presence of ApoE4 has been shown to impair the integrity of the blood-brain barrier.Research has shown that individuals carrying theApoEε4gene have higher levels of ferritin in their cerebrospinal fluid than non-carriers.Furthermore, there is a significant positive correlation between the concentration of ApoE protein and the level of ferritin in the cerebrospinal fluid (Ayton et al., 2015).A separate study found that individuals with AD who had high levels of ferritin in their cerebrospinal fluid exhibited more pronounced hippocampal atrophy and lateral ventricle enlargement; furthermore, amyloid deposition was elevated with increasing concentrations of ferritin (Goozee et al., 2018).ApoE is a crucial protein that plays an important role in regulating lipid metabolism in the central nervous system.ApoE exerts functionality by inhibiting the synthesis of enzymes in the cholesterol synthesis pathway; this leads to the accumulation of acetyl-CoA,a precursor of cholesterol and PUFA synthesis.The accumulation of acetyl-CoA creates an environment that promotes LPO and ferroptosis (Li et al.,2021).ApoE4 is generated by astrocytes, which can substantially amplify the inflammatory response and contribute to the production of Aβ.However, this process negatively impacts the elimination of Aβ by astrocytes, thus leading to the excessive phosphorylation of tau protein.This process ultimately results in the decline and loss of nerve function, and eventually neuronal death (Shi et al., 2017; Li et al., 2021).These results indicated that APOE may contribute to the development of AD by inducing ferroptosis and damaging the blood-brain barrier.However, additional experiments are now necessary to substantiate this hypothesis.Furthermore, the precise pathogenic mechanism underlying AD remains unclear, although various hypotheses have been proposed previously.It is possible that ferroptosis affects the onset and progression of AD by a range of mechanisms, thus necessitating further research.Consequently, there are significant research opportunities in the field of AD.It is clear from the existing literature that ferroptosis has introduced novel concepts for understanding the development of AD and has the potential for targeted therapy.However, further research is needed to determine if this knowledge can be successfully translated into clinical practice to improve the survival rates of AD patients and enhance their quality of life.
Acknowledgments:The authors thank Tianyi Zhuang from Faw-Volkswagen Automotive Co.,Ltd.for assistance with language edit.
Author contributions:Conceptualization and supervision:BS;literature collection:LX,QS,YH,JS,LZ;manuscript draft:LF,CF,ML;figure preparation:CF,ML.All authors approved the final version of the manuscript.
Conflicts of interest:None declared.
Data availability statement:Not applicable.
Open access statement:This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Brian Zhiyang Wang,National Neuroscience Institute,Singapore.
Additional file:Open peer review report 1.
杂志排行

中国神经再生研究(英文版)的其它文章
- The big data challenge – and how polypharmacology supports the translation from pre-clinical research into clinical use against neurodegenerative diseases and beyond
- P-aminobenzoic acid promotes retinal regeneration through activation of Ascl1a in zebrafish
- Lupenone improves motor dysfunction in spinal cord injury mice through inhibiting the inflammasome activation and pyroptosis in microglia via the nuclear factor kappa B pathway
- Two-photon live imaging of direct glia-to-neuron conversion in the mouse cortex
- Neutrophil extracellular traps mediate neuroimmunothrombosis
- Impact of increasing one-carbon metabolites on traumatic brain injury outcome using pre-clinical models