雷公藤红素通过调控ATF4/caspase-3/GSDME信号通路促进肝癌细胞焦亡
2024-02-04王永辉孔永红
李 端,王永辉,周 静,张 乐,孔永红
(1.黄淮学院直属附属驻马店市中心医院药学部,河南 驻马店 463000;2.苏州大学药学院药理学系,江苏 苏州 215123;3.黄淮学院化学与制药工程学院,河南 驻马店 463003)
肝细胞癌(hepatocellular carcinoma, HCC)是全球最常见的人类致命恶性肿瘤之一,其中以原发性肝癌占比最高,约占85%。据统计,HCC每年约有 840 000新发病例和 780 000例死亡病例[1]。大多数肝癌患者早期无症状确诊时已发展到晚期,往往失去了手术治疗的最佳时机。靶向治疗是这些HCC患者的重要选择,大量研究证实,靶向治疗可以在一定程度上改善晚期 HCC患者的生存率[2]。但靶向药物发展缓慢,临床可用于HCC治疗的靶向药物极为有限且易产生耐药。因此,有迫切需要开发新的和更具体的治疗HCC的方法。近年来,中药因其作用广、副作用小、协同治疗效果好等多方面优点成为抗肿瘤治疗的“热点”。雷公藤红素(tripterine, TRI)是一种天然的化合物,主要来源于雷公藤。TRI已被证明具有抗炎、抗氧化、抗癌和抗纤维化的作用。在临床上,TRI已被用于治疗类风湿性关节炎、麻风反应和其他自身免疫性[3-6]。TRI能有效通过诱导线粒体损伤来对抗结肠癌细胞和细胞内钙的积累。TRI还能通过促进线粒体活性氧(reactive oxygen species, ROS)产生,诱发细胞线粒体膜电位 (mitochondrial membrane potential, MMP) 和DNA损伤,从而改善急性淋巴细胞白血病[7]。也有研究报道,TRI能抑制 HCC 细胞的生长和诱导 HepG2 细胞凋亡[8]。这些研究表明TRI可能是一种潜在的癌症治疗药物。然而,TRI抗癌作用的分子机制仍有待阐明。细胞焦亡是一种新的程序性细胞死亡形式,其特征是细胞肿胀并伴质膜表面孔隙。细胞焦亡最初归因于半胱天冬酶(caspase)对gasdermin D (GSDMD)的蛋白水解片段化。最近的一些研究表明了一种全新的Gasdermin 家族的另一个成员gasdermin E (GSDME), 其可以诱导细胞凋亡向细胞焦亡转换[9]。与GSDMD 类似,GSDME 包含 N 端和 C 端结构域,N 端单体在等离子体中低聚形成孔膜[10]。活化的 caspase-3酶剪切 GSDME域间链接器释放 N 端域并形成细胞膜孔导致细胞焦亡,最终导致细胞乳酸脱氢酶(lactate dehydrogenase, LDH)活性升高以及碘化丙啶 (PI) 摄取的增加。本研究探讨TRI是否可通过调节ATF4/caspase-3/GSDME信号通路诱导肝癌细胞焦亡。
1 材料和方法
1.1 主要试剂TRI(纯度>98%,购自成都普菲德生物技术有限公司,批号20210416)。
1.2 细胞系和细胞培养HepG2 细胞(人 HCC 细胞系)和 Hepa1-6 细胞(小鼠HCC细胞系)购自上海细胞生物研究所(中国上海)。HepG2和Hepa1-6 细胞分别在MEM和 DMEM培养基中培养。所有的培养基都是含10%胎牛血清(FBS;赛默飞世尔Scientific, Waltham, MA, USA)、青霉素 (100 kU·L-1) 和链霉素(100 g·L-1;Hyclone, Logan, UT, USA)。细胞在 37 ℃和5% CO2下培养。GSDME质粒由 GenePharma(中国上海)合成。通过蛋白质印迹法验证构建是否成功。m-GSDME forward, 5′-CTGCGGATGACTGCCTCAATGG-3′,h-GSDME reverse, 5′-ATTGCTTGTGCTGTGCGTTGC-3′; h-GSDME forward, 5′-CCTGTGGCATCCACGAAACT-3′,h-GSDME reverse, 5′-GAAGCATTTGCGGTGGACGAT-3′。转染前,将2 mL含有1.0×106细胞的完全培养基放入6孔板。待细胞汇合度达到70%左右后,每个细胞中的完全培养基用1.8 mL不含FBS的基础培养基代替。然后2 μg模拟物或质粒分别用 100 μL Opti-MEM (31985070, Gbico) 稀释。此外,2 μL lipo 3000 (BMB1385, BOMEI BIOTECHNOLOGY,合肥) 也用100 μL Opti-MEM稀释。然后将稀释的模拟物或质粒和稀释的 lipo 3000充分混合。孵育10 min后,最后将混合溶液加入每个孔,进一步培养 48 h用于后续分析。
1.3 细胞活力测定和LDH释放测定接种指数生长的 HepG2 细胞或 Hepa1-6 细胞在刺激前 24 h放入96孔板(7 000 个细胞/孔)中。将TRI溶解在 DMSO 中并稀释用不同浓度的培养基 (DMSO终浓度<0.1%)。用TRI孵育指定的时间和浓度。通过CCK-8测定细胞活力(Dojindo 实验室,日本九州岛)。根据Prism 6计算的的细胞活力值计算半数最大抑制浓度(IC50)。对于LDH释放,细胞培养上清液收集后,使用LDH 检测试剂盒(Beyotime 生物技术研究所),在黑暗室温下孵育30 min。在酶标仪(Thermo Scientific Varioskan LUX,Waltham,MA,USA)450 nm 处测定吸光度分光光度。
1.4 免疫荧光用于免疫荧光染色的细胞用4%多聚甲醛固定 20 min,然后使用 0.5% Triton X-100渗透15 min。将细胞在 3% BSA中孵育30 min。之后,细胞孵育一抗并4 ℃过夜。PBS 3 次洗涤后,孵育二抗(Invitrogen,美国)2 h。最后,将印迹用DAPI染色5 min(Beyotime),在荧光显微镜下观察细胞(徕卡,德国)。
1.5 通过流式细胞术评估细胞凋亡收集细胞,根据说明书,用PBS洗涤2次并使用FITC和PI染色(益生,中国)。染色的细胞是用流式细胞仪检测,并使用FlowJo软件进行数据处理。
1.6 蛋白质印迹收集细胞总蛋白,并BCA 试剂盒(QPBCA,默克)测定其浓度。然后,使用SDS-PAGE 凝胶对30 μg 总蛋白和4 μL标记物(PR1910,Solarbio,中国)进行电泳,随后转移到0.22 μm PVDF膜(W015-2,建城生物,中国)。用5%脱脂牛奶封闭后,膜与下列相关一抗在4 ℃孵育12 h:抗人 GSDMD (1 ∶1 000, Proteintech),抗 GSDME (1 ∶1 000, Abcam),抗兔半胱天冬酶3 (caspase3,1 ∶1 000)、抗兔聚 ADP-核糖聚合酶 (PARP, 1 ∶1 000)、抗鼠 GSDMD(1 ∶1 000,Abcam)和抗甘油醛-3-磷酸脱氢酶(GAPDH, 1 ∶5 000)均购自Cell Signaling Technology。抗兔P-eIF2α (1 ∶1 000),抗兔eIF2α (1 ∶1 000),抗兔ATF4 (1 ∶1 000) 均购自Abcam。然后,2 h后与兔二抗(1 ∶10 000,Abcam)或鼠二抗孵育抗体(1 ∶10 000,Abcam),进一步用200 μL ECL发光液进行显影(P0019,Beyotime,上海)。最后,检测到信号图像使用IPP6.0软件进行分析。
1.7 动物研究40只雄性BALB/c裸鼠(6周,体质量18~20 g)获购自扬州大学比较医学中心(扬州,中国)并保存在动物实验中心中国药科大学。随机分为4组,阴性对照 (NC)、si-GSDME组、TRI组和TRI+si-GSDME组,NC和TRI组给予未干扰的Hepa1-6细胞3×106被皮下注射到小鼠右上肢腋下;si-GSDME组和TRI+si-GSDME组小鼠接种等数量si-GSDME干扰的Hepa1-6 细胞。每3 d监测一次小鼠肿瘤体积(长×宽×宽×3.14/6)和体质量。在治疗结束时,处死小鼠,取出肿瘤,称质量。动物实验通过本机构动物伦理委员会批准。
1.8 组织病理学检查收获小鼠的肿瘤组织和肝脏,并用4%多聚甲醛固定,脱水,包埋石蜡。
2 结果
2.1 TRI抑制HCC细胞增殖和侵袭TRI在各种癌症中发挥抗肿瘤作用,但在肝癌中的研究有限[7-8]。因此,我们考察了TRI是否可以抑制HCC细胞增殖。CCK-8结果表明,TRI可呈浓度和时间依赖性抑制肝癌细胞HepG2、Hepa1-6的增殖(Fig 1A和1D)。细胞实验结果显示,随着TRI浓度的增加,肝癌细胞HepG2、Hepa1-6的克隆数均明显减少(Fig 1B和1E)。同时,细胞Transwell实验结果表明,随着TRI浓度的增加,肝癌细胞HepG2、Hepa1-6的侵袭(Fig 1C和1F)能力。
2.2 GSDME对TRI诱导的肝癌细胞焦亡至关重要据报道,TRI可通过诱导肿瘤细胞凋亡抑制肿瘤进展[8]。我们试图确定是否还可通过其他方式抑制 HCC 进展。有趣的是,在TRI处理后的 HepG2 和Hepa1-6 细胞均出现了焦亡(Fig 2A)。为了证实这一发现,我们评估了焦亡相关蛋白。在gasdermin 蛋白家族成员中, GSDMD 的裂解可以将 GSDMD 的 N 端结构域释放到膜上。虽然 GSDMD 在 HepG2和 Hepa1-6 细胞中表达,但TRI处理不能诱导GSDMD分裂 (Fig 2B)。此外,在Gsdmd siRNA的Hepa1-6(Fig 2C)不影响TRI诱导的细胞死亡和 LDH 释放(Fig 2D 和E)。进一步我们检测了新细胞焦亡效应因子GSDME的表达,结果表明,TRI呈剂量依赖性方式诱导HCC 细胞 caspase 3和PARP裂解(Fig 2F-I)和 N 端 GSDME (N-GSDME)的释放。

Fig 1 Proliferation and invasion of HCC cells inhibited by TRI
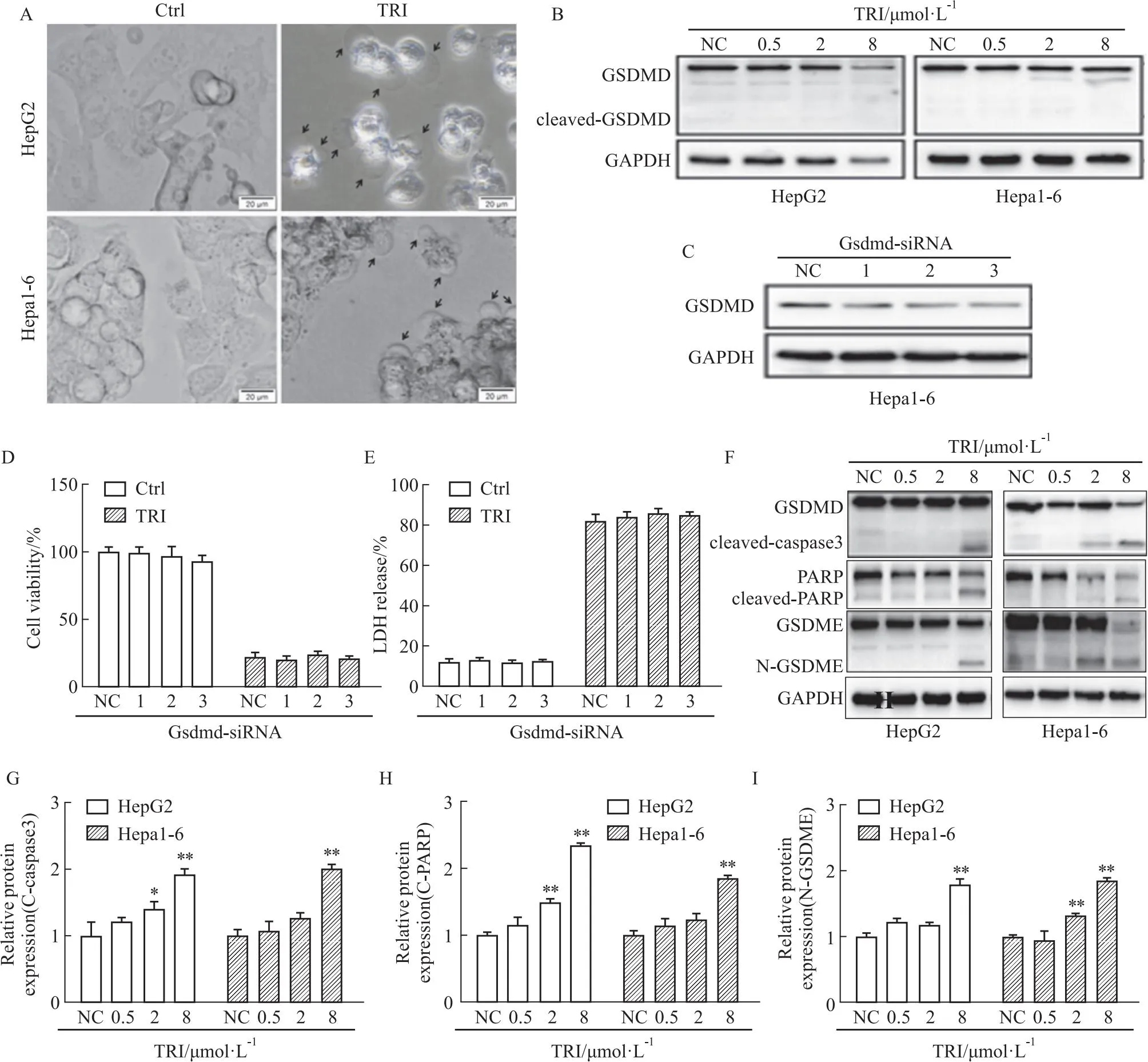
Fig 2 TRI-induced pyroptosis of hepatoma cells

Fig 3 GSDME mediated response of HCC cells to TRI
2.3 GSDME缺失可部分逆转 TRI 对肝癌细胞抑制作用为了验证GSDME的作用,我们沉默Hepa1-6 细胞中的 GSDME(Fig 3A-F)。结果显示,GSDME缺失明显减少TRI诱导的 LDH 释放和质膜膨胀,虽然没有挽救细胞死亡。同时,GSDME缺失后可部分逆转细胞TRI抑制肝癌细胞菌落形成和迁移的作用(Fig 4A-D)。TRI处理的Hepa1-6细胞迅速进入膜联蛋白V(annexin V)和PI双阳性阶段,而Gsdme的敲低后随着annexin V单阳性细胞比例的增加和双阳性细胞百分比的降低而延迟细胞的凋亡(Fig 4E-F)。这些发现表明TRI发挥其作用部分取决于HCC细胞中的GSDME。
2.4 TRI激活 eIF2α/ATF4轴触发GSDME介导的细胞焦亡为进一步阐明细胞焦亡的发生机制,我们检测了eIF2a/ATF4轴,这已被证明是caspase-3的上游调控通路。Western blot结果表明,TRI刺激后HepG2 和 Hepa1-6 细胞中 p-eIF2a、ATF4均显著升高(Fig 5A-C)。同时,免疫荧光结果也显示,TRI刺激后HepG2 和 Hepa1-6 细胞中ATF4阳性细胞数明显增加,并呈剂量依赖性(Fig 5D,E)。
2.5 TRI抑制肿瘤生长部分依赖于GSDME进一步研究 TRI的抗HCC肿瘤作用,我们进行了体内动物研究。结果显示,TRI治疗显着抑制肿瘤生长、减少肿瘤重量,且TRI+si-GSDME 敲低组肿瘤增长明显快于TRI+NC组。(Fig 6A-C)。总之,TRI抑制肿瘤生长部分依赖于GSDME。

Fig 4 GSDME mediated response of HCC cells to TRI
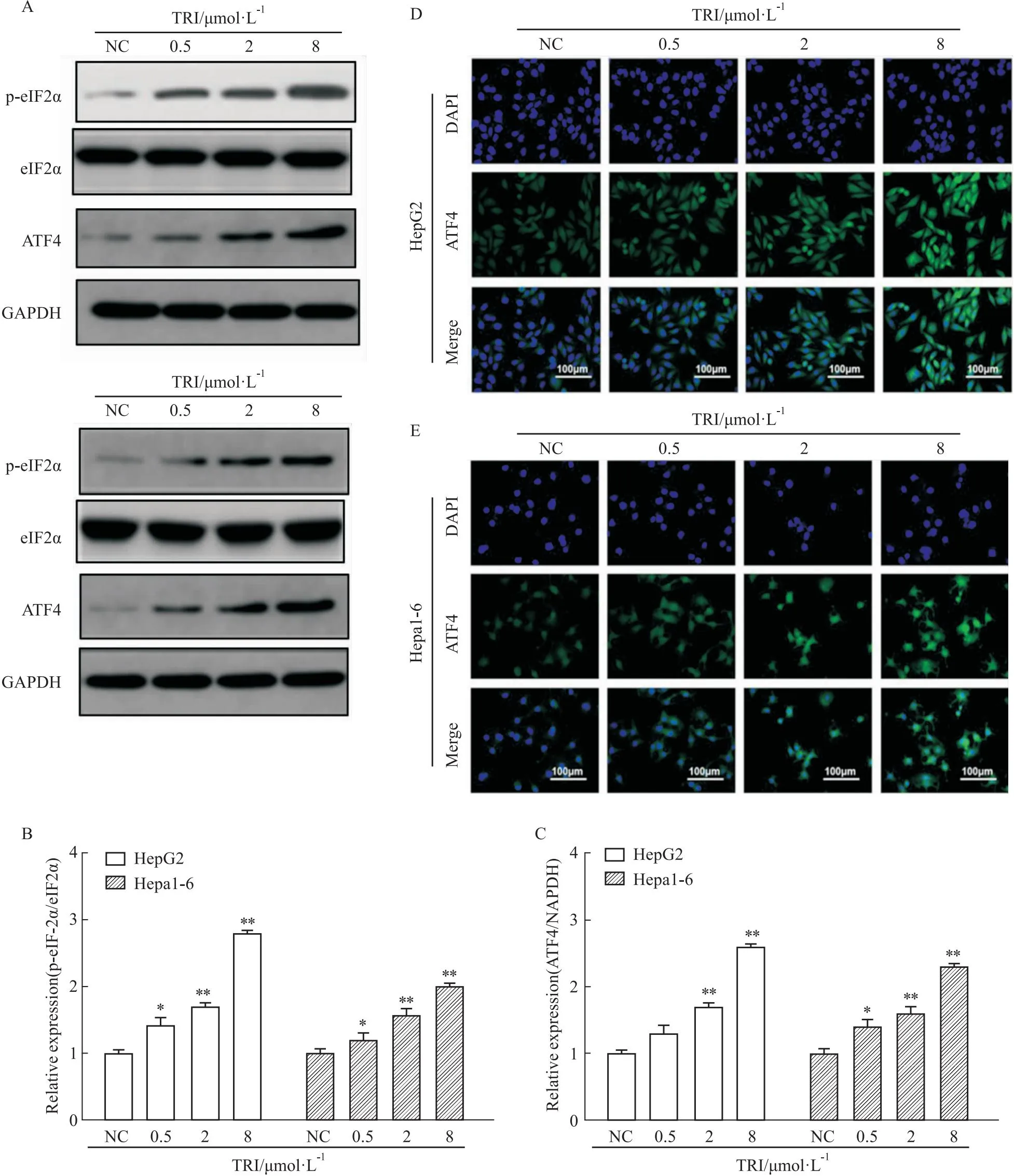
Fig 5 TRI activation of eIF2α/ATF4 axis triggered GSDME-mediated pyroptosis

Fig 6 TRI inhibited growth of HCC cells in vivo
3 讨论
HCC总体 5年生存率仍然很差低。化疗仍然是晚期 HCC的主要治疗方法。索拉非尼是晚期 HCC 患者的标准疗法,但它只能为患者提供有限的生存益处[11]。所以,迫切需要确定更有前途的高活性且低毒性的HCC治疗药物。细胞活力实验表明,TRI可以明显抑制HepG2或 Hepa1-6 细胞生长。此外,体内实验HCC 模型显示,TRI治疗抑制HCC肿瘤的生长。
细胞凋亡通常被认为是调节肿瘤细胞死亡的主要形式。TRI用于癌症治疗,也归因于其抑制癌细胞的生长能力并诱导癌细胞凋亡。已有研究发现TRI可诱导HepG2细胞凋亡[12]。在本研究中,我们扩展了传统观点,并提出 GSDME 依赖性细胞焦亡参与TRI在 HCC 中的治疗,这一观点得到了以下实验数据验证。首先,在TRI处理的 HCC 细胞中观察到细胞焦亡现象,如细胞膜裂解、气球样气泡、LDH释放和 PI 阳性染色。但值得注意的是,经历坏死性凋亡的细胞也表现出质膜透化,细胞肿胀和溶解。在细胞焦亡期间,焦亡发生时形成孔隙,它允许细胞质的内容物,如LDH和炎性细胞因子释放,荧光标记的膜联蛋白V (Annexin V)、7-氨基放线菌D (7-AAD)或PI可进入细胞。相比之下,膜不透性染料,如作为7-AAD或 PI,可通过孔隙进入使焦亡细胞着色,但不染色凋亡细胞。我们发现TRI处理的 Hepa1-6 细胞LDH水平明显升高,且Annexin V 和 PI 双阳性细胞比例增多,提示TRI诱导了肝癌细胞焦亡。同时,肝癌细胞GSDME 沉默后没有挽救TRI诱导的肝癌细胞死亡,而减少了TRI诱导的 LDH 释放和细胞肿胀,并减少了双阳性细胞的百分比。以上表明,TRI诱导肝癌细胞焦亡可能与激活GSDME表达有关。GSDMD是细胞焦亡通路代表性蛋白,已有大量研究证明,多种中药可通过caspase-1/GSDMD通路抑制肿瘤的发生[13-15]。本研究Western blot结果显示,TRI作用于肝癌细胞后,细胞中N-GSDMD蛋白水平未明显改变,且击倒Hepa1-6细胞的 Gsdmd 没有影响TRI诱导的细胞死亡和 LDH 释放。因此,GSDMD 不参与TRI诱导的 HCC 细胞焦亡,主要是GSDME。GSDME 依赖性细胞焦亡可由促凋亡蛋白caspase-3触发。我们发现TRI诱导Hepa1-6 细胞中cleaved caspase-3、cleaved-PARP和 N-GSDME 的蛋白质水平呈剂量依赖性增加。因此,TRI可以诱导通过调控 caspase-3/GSDME 途径诱发肝癌细胞焦亡。但TRI是如何激活caspase-3/GSDME的呢?
eIF2α/ATF4/CHOP通路是一个重要的信号通路调节细胞存活的途径。磷酸化真核起始因子 2 α (eIF2α) 的表达受到刺激后,可增强激活转录因子 4 (ATF4),依赖于 ATF4-C/EBP 同源蛋白 (CHOP) 的表达上调,促进细胞凋亡[16]。据报道,eIF2α 通过多种途径诱导细胞凋亡,包括线粒体依赖性途径、死亡受体途径和其他途径[17]。研究表明,eIF2α在细胞凋亡中起关键作用并且与 ER 应激引起的许多疾病有关。在正常情况下,p-eIF2α的表达较低。然而,在病理情况下,p-eIF2α会急剧表达并引发细胞凋亡[18]。同样,不同剂量TRI对肝癌细胞中p-eIF2α表达的影响,随着TRI剂量的增加,p-eIF2α蛋白的水平明显增加。最近的研究表明,化疗药物诱导的细胞焦亡是由 BAK/BAX-caspase-3-GSDME介导的,表明 BCL-2 家族的促凋亡因子可能参与细胞焦亡。此外,线粒体功能障碍触发 caspase-3-GSDME 通路激活介导米替隆诱导的肝癌细胞焦亡。因此可能会有类似的诱导细胞焦亡的机制。以前的研究表明,ATF4/CHOP 可能是调节 BCL-2 家族的关键上游转录因子[19]。本研究显示,TRI作用的肝癌细胞中ATF4蛋白水平明显升高。这些结果表明 eIF2α/ATF4/caspase-3 通路是TRI 诱导GSDME升高的原因。最后,体内证据进一步证明TRI在 HCC 肿瘤中诱导肿瘤细胞焦亡。
综上,TRI可能是一种潜在的治疗HCC的候选药物,并指出ATF4/caspase-3/GSDME可调节TRI诱导的焦亡过程。
