小清蛋白阳性抑制性中间神经元与视觉发育可塑性的研究进展△
2024-01-30杨诗巧
杨诗巧 郝 瑞 张 伟
目前研究发现,大脑皮层主要存在谷氨酸能兴奋性投射神经元和γ-氨基丁酸(GABA)能抑制性中间神经元(INs)[1]。GABA-INs在大脑皮层中的占比相对较少,在人脑皮层中仅占30%~45%,在猕猴脑皮层中占20%~25%[2],在小鼠和大鼠脑皮层中占20%~30%[3-5], 但由于其短树突的形态特点,主要产生局部神经回路,故对大脑皮层中兴奋性/抑制性网络平衡的维持至关重要。
INs根据其表达分子不同分为四种类型:小清蛋白(PV) 阳性、生长抑素(SST)阳性、血管活性肠肽阳性以及卷轴蛋白阳性INs[6]。其中PV+INs和SST+INs同起源于腹侧端脑皮质下区内侧神经节隆起(MGE)和视下区[3-7]。本文就PV+INs在视皮层发育中的作用进行综述,以期能够为视皮层可塑性研究提供一定的指导。
1 PV+中间神经元的发育和分型
人胚胎孕第6周开始出现神经节隆起,此时中间神经元祖细胞向皮层迁移;至孕8周,MGE初步形成;孕14周时,神经节隆起区中间神经元祖细胞大量产生;在孕8~14周时MGE表达转录因子LHX6的细胞切线迁移至皮层[8-9],该转录因子激活依赖于NKX2-1,而NKX2-1表达受上游音猬因子通路调节,其下游转录因子SOX6表达促进PV+INs的成熟及最终定位[8,10];孕16~20周,不同亚型中间神经元在皮层区域大量出现,而不同的转录因子表达SST、PV、神经肽Y、钙网膜蛋白或钙结合蛋白促进这一过程发生。研究发现,PV+INs和SST+INs的产生并非同时进行,孕22 d小鼠,SST先于PV产生,此过程受核素受体TFs CoupTF2调控,E11.5 d(即胚胎期 11.5 d)和E14.5 d时CoupTF2缺失引起出生后PV+INs增多,而E12.5 d时该受体功能的缺失则导致出生后SST+INs减少,从而表明CoupTF2在小鼠E11.5 d和E14.5 d时抑制MGE的中间前体细胞向PV+INs的分化[11-12],但是否存在其他核素受体调控PV+INs和SST+INs分化和发育尚待进一步研究。此外,神经营养因子、胶质源性神经营养因子等均在PV+中间神经元迁移中起精细调控作用[13-14]。
PV+INs依据细胞形态分为两种亚型:篮状细胞和吊灯状细胞;前者出现较早,后者的功能目前存在争议。有研究显示,吊灯状细胞对靶向的锥体神经元产生兴奋效应[15]。INs中特异性PV的表达在出生后开始,目前尚未明确早期PV+INs的特征性标志物[14],因此,控制PV+INs向篮状细胞和吊灯状细胞分化的机制亦有待进一步研究。
2 PV+INs与视皮层可塑性及视觉发育关键期的关系
PV+INs在控制锥体细胞兴奋、维持神经网络平衡以及生物节律等方面起着重要作用。出生后随着视觉发育及周围环境的刺激,以及突触增多,PV+INs 逐渐成熟,神经网络稳态也随之建立。其主要分布在大脑新皮层的L5/6,在L1中几乎没有。PV对钙离子和镁离子具有高亲和性,在动作电位后可使细胞回到静息电位,从而促进PV+INs的成熟[16]。而PV+INs本身动作电位的激发是连续或延迟的,单个动作电位的激发具有快尖峰动力特征,具有较小或者中等大小的输入阻抗以及较大的超极化后电位[1,7],这样的电生理特征使其突触反应十分迅速,也表示了其代谢高且易受氧化应激损伤的特质。研究表明,PV+INs在快速动眼睡眠时活性增加,且在维持大脑清醒状态中起重要作用,这主要与成熟的PV+INs形成稳定神经网络和γ振荡有关[17-18]。PV+INs主要投射在锥体神经元的胞体和近端树突,相比其他INs,其与锥体神经元距离最近,具有更强的抑制性支配。PV+INs对其他INs几乎没有抑制作用,但被SST+为主的其他INs抑制性支配[19]。PV+INs参与视皮层可塑性调控的相关机制见图1。
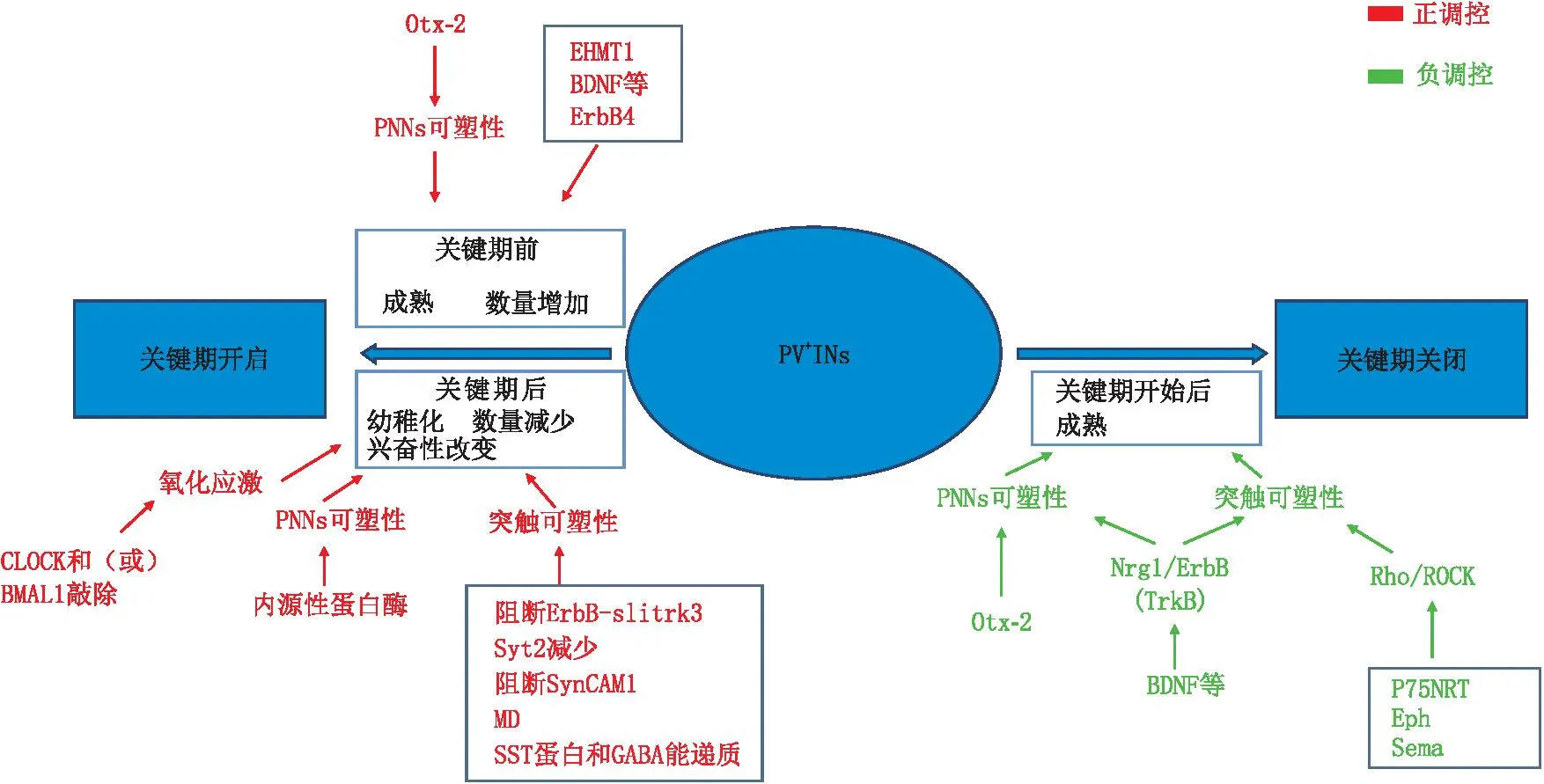
PNNs:周围神经网;BDNF:脑源性神经营养因子;ErbB:受体蛋白酪氨酸激酶;TrkB:酪氨酸激酶;P75NRT:神经生长因子受体;Eph:蛋白酪氨酸激酶亚家族;Sema:信号素。图1 PV+INs参与视皮层可塑性调控机制图
2.1 PV+INs-未成熟的周围神经网稳固状态的破坏
正常小鼠出生后第2周,视皮质中开始表达PV,PV+INs也在此时出现较为稳定的固有膜特性(膜电阻、膜电容和静息电位),同时,未成熟的周围神经网(PNNs),即视皮层内主要包裹PV+INs的细胞外基质成分被首次观察到[20];出生后第3周时,其数量及增长速度渐趋稳定[21](图2)。猫视皮层的研究进一步表明,PV+INs数量在关键期后仍继续增长,而PNNs比PV+INs发育晚两周,但成年后二者数量基本稳定,PV+INs数量占皮层神经细胞数量的10%~20%,占全部INs总数的40%左右[22-23]。研究表明,部分PV+INs的成熟触发了关键期的开启,至关键期结束,70%PV+INs周围包裹着PNNs,85%的PNNs包裹着PV+INs[24],此时PV+INs的抑制作用也达到最大[25-26]。Xia等[27]认为这种变化是由于成熟的PNNs抑制了PV的表达导致的。视皮层可塑性也随着关键期的结束维持在低水平。Carstens等[26]认为,成熟的PNNs抑制神经元轴突生长以及作为屏障阻断某些神经递质而发挥限制可塑性的功能,通过消除PNNs能够重新恢复成人视皮层可塑性,同时观察到PV+INs动作电位发生变化。

图2 PV+INs和PNNs发育趋势图
PV+INs的幼稚化能够触发关键期开启[26],其特征性改变为PNNs结构完整性破坏和皮质中γ振荡降低或消失[28],PNNs使其包裹的PV+INs轴突生长受限。其中发挥作用的因子是同源蛋白Otx2,一种能够促进PV+INs成熟的蛋白,从而加速关键期关闭。而Otx2-AA型小鼠(Otx2结合位点突变)幼年关键期出现延迟,表明Otx2对可塑性的调控是双向的, Otx2产生于视网膜,而视皮层中的PV+INs能够优先募集Otx2离不开PNNs上的特异性复合糖,短暂的PNNs消除实验中无法观察到PV+INs电生理的改变,只有当Otx2浓度下降时,PV+INs电生理特性才随之改变,此时可塑性发生改变[27]。因而,成年期PV+INs-PNNs的互相稳固离不开Otx2的介导[29],目前具体的结合位点仍在研究。同时敲除四种组成PNNs的硫酸软骨素蛋白多糖可以引起仅限于V1的PV+INs数量减少,但Otx2仍能被PV+INs内化,可能是由于残余的PNNs中存在Otx2的结合位点[30]。
目前提出一种PNNs成分蛋白聚糖可能为其作用位点[25]。除了Otx2,关键期长达10 d的暗饲养能够降低蛋白聚糖浓度,从而减少视皮层PNNs含量,包裹PNNs的PV+INs体积缩减了19%,但数量几乎不受影响,而未包裹PNNs的PV+INs体积缩减了17%,二者没有显著差异[22]。体积变小可能是由于细胞膜蛋白聚糖的缺失, PV+INs体积的改变在可塑性变化中发挥的作用有待进一步研究,但可以肯定的是,PNNs本身完整性的破坏不会直接引起可塑性的改变,其上一些复合糖才是关键期的开关,例如蛋白聚糖的裂解促进皮质发生可塑性变化,PNNs的降解联合感觉剥夺才能引起可塑性的改变证实这一点[23]。研究发现,内源性蛋白酶对蛋白聚糖有直接靶向作用,PV+INs作为体内丝氨酸类组织型纤溶酶原激活物(tPA)的主要来源之一,通过细胞自主纤溶酶原依赖途径降解包裹在周围的PNNs,条件性敲除PV+INs的tPA后,PNNs密度增加[31],病理状态下tPA-纤溶酶系统参与皮质可塑性[32],目前激发PV+INs释放tPA的因子尚待探索,而其他的内源性蛋白酶可作为未来研究方向。简言之,PV+INs在特定条件下释放内源性蛋白酶tPA,激活纤溶酶原系统作用于PNNs的蛋白聚糖,继而降解PNNs。考虑到蛋白聚糖也是Otx-2的作用位点,tPA降解PNNs可能减少Otx-2的募集从而恢复皮质可塑性。
除了Otx-2和tPA,神经营养因子参与关键期可塑性也被认为和蛋白聚糖有关[33]。神经营养因子是一类神经元生长发育所必需的蛋白质,主要包括脑源性神经营养因子(BDNF)、神经营养因子-4、胶质细胞源性神经营养因子等。其中BDNF及其受体蛋白酪氨酸激酶(ErbB)、酪氨酸激酶2(Ntrk2,TrkB)在诱导PV+INs成熟方面的作用已被证实,依赖于经典Nrg1/ErbB(TrkB)信号通路,该信号通路表达下调可以观察到成年小鼠中视皮层具有幼年可塑性[34],而蛋白聚糖促进这一通路表达,意味着限制视皮层可塑性。
PV+INs表观遗传学修饰也能影响可塑性。Rett综合征为一种甲基-cpg结合蛋白2(Mecp2)突变的X连锁基因遗传病,主要表现为严重的神经发育障碍,在PV+INs中条件性敲除Mecp2的小鼠(PV-Mecp2-/y)不会产生Rett综合征的大多行为,但在出生后26~30 d眼优势可塑性完全消失[35],表明PV+INs的去甲基化导致小鼠原本的关键期可塑性消失。Patrizi等[36]研究显示,PV+INs Mecp2条件性敲除的小鼠在出生后15 d开始显现出PV+INs的增多,伴随着成熟型PNNs、更大的自发抑制电流的出现,这有利于抑制性网络形成,过早出现眼优势转移。因而,推论出在PV+INs特异性敲除Mecp2小鼠关键期提前使其在出生后26~30 d期间而单眼剥夺无法诱导出眼优势转移。
总之,PV、PV+INs和PNNs的数量与关键期的可塑性变化趋势呈负相关,从而表明:(1)PV+INs的成熟是经验依赖性的;(2) PV+INs控制着视觉发育关键期的开始和关闭。至此,抑制性回路形成,突触结构稳定,视皮层神经网络中兴奋-抑制达到平衡,关键期结束,视皮层可塑性被限制;反之,其中任何一个因素受到干扰发生改变,都会导致关键期开启。
2.2 PV+INs通过锥体神经元影响突触可塑性
大脑皮层中PV+INs与锥体神经元通过突触形成前馈抑制和反馈抑制。局部兴奋性突触机制的研究表明,PV+INs与锥体神经元之间兴奋性回路的形成依赖于经典的Nrg1/ErbB4信号通路,其活性变化受Nrg1浓度影响,而该信号通路的高表达是视觉发育关键期的“刹车”[13,37],但亦有研究发现其高表达只是限制了成人可塑性,将皮质可塑性维持在极低水平,Nrg1抑制剂可诱导眼优势转移,改善弱视成年期大鼠视力[37-38],但PV+INs中的Nrg1来源尚不明确。
不同于兴奋性突触,ErbB4介导的锥体神经元抑制性突触形成不依赖于酪氨酸激酶依赖的经典通路,其作为细胞黏附分子与抑制性突触后膜特异性标志物slitrk3相互作用,阻断该过程可导致抑制性突触减少,从而使得抑制回路形成缺陷[39]。Winkel等[40]提出酪氨酸激酶TrkB的光激活降低了PV+INs兴奋性,引起抑制性突触标志物突触结合蛋白2Syt2的表达减少,降低了前馈抑制作用,从而激活可塑性。相较于局部锥体神经元,Kloc等[41]提出来自丘脑外侧膝状体的长程投射神经元更强烈地激活 PV+INs,而Ribic等[42]则提出一种细胞黏附分子SynCAM1介导该兴奋性突触形成,阻断该分子作用同样可以再激活成年视皮层可塑性。
以上均是在视皮层L4中观察到的结果,而单眼剥夺能够使L2/3的锥体神经元与PV+INs间兴奋性突触短暂失活,这种失活表现为全或无[43],但不同于L4中Nrg1的作用,其失活是由神经中枢蛋白2介导。单眼剥夺引起的PV+INs短暂的突触失活导致的抑制作用下降被认为是后期皮质发生可塑性变化的基础[44]。
此外,细胞骨架动力学说表明,p75NTR上调激活Rho/ROCK信号通路,诱导细胞骨架重组、细胞迁移和应力纤维形成,抑制PV+INs轴突生长和突触形成,发挥类似作用的还有促红细胞生产素和脑信号蛋白信号素[45]。
2.3 PV+INs本身与视皮层可塑性的联系
一定浓度的PV和PV+INs的成熟是启动关键期的重要因素,主要原因可能是PV+INs本身膜电阻以及静息膜电位的改变,也就是内在可塑性发生变化[45],关键期前成熟PV+INs数量减少必然会影响关键期开启。 EHMT1基因敲除的小鼠,其睁眼时间推迟且V1中PV+INs成熟延迟,从而引起关键期开始延迟,并且可持续到出生后100 d[46]。上述提到BDNF等通过TrkB激活下游PI3激酶信号通路。此信号可能由Rho GTPase家族介导作用于肌动蛋白,从而强烈刺激MGE起源的中间神经元的切向迁移,早期发育时BDNF的浓度降低会导致包括PV+在内的中间神经元数量减少[34]。另一种观点认为,ErbB4在中间神经元向皮层发育迁移的过程中发挥十分重要的作用,其敲除导致成年小鼠中INs的数量显著下降,但不依赖于ErbB4活性[46],此外是否存在其他机制尚不明确。
PNNs对PV+INs有保护作用,PV作为一种钙结合蛋白对细胞内Ca2+有亲和作用,作为钙离子缓冲剂介导细胞恢复静息电位,其缺失导致细胞内Ca2+病理性堆积,从而引起PNNs降解以及PV浓度降低,机体对氧化应激损害敏感[16]。在敲除生物钟基因CLOCK和(或)BMAL1的大鼠中观察到CLOCK/ BMAL1复合体的缺失,抑制了上游基因表达和细胞内cAMP/蛋白激酶A信号通路,导致氧化物堆积,诱导PV+INs的损伤[47]。上述PV+INs的缺失多在认知功能障碍疾病中被证实,其诱导的认知功能减退和皮层幼稚化不仅和神经网络兴奋/抑制失衡息息相关,其与视觉系统可塑性的关联性值得我们探究。
3 探索PV与其他类型神经元对视皮层可塑性的影响
SST+INs与PV+INs同起源于MGE,且数量达GABA-INs的20%~30%,许多免疫组织化学染色结果显示,SST+INs分泌的GABA能神经递质和SST蛋白少数直接作用于PV+INs的胞体和近端树突,而大部分以膜接触的形式传递信号,减少PV+INs上的兴奋性突触来降低抑制性,提高V1锥体神经元兴奋性,改变视皮层神经元活动以及神经网络稳定性[48]。
除了中间神经元,大脑皮质中另一类神经细胞——胶质细胞也参与视皮层可塑性的变化。其中,在重复麻醉剂量氯胺酮恢复小鼠视皮层可塑性的研究中,证实了反应性小胶质细胞和PV+INs相互作用,用药后短时间内可观察到反应性小胶质细胞吞噬PNNs片段,这个过程可能通过MMP-9分解PNNs[28],三者之间的具体作用机制仍待研究。
4 基于PV+INs与视皮层可塑性的弱视治疗方法
弱视是一种由于双眼信息输入不平衡导致的疾病,通常伴有大脑视皮层结构和功能改变,关键期视皮层可塑性的存在有利于纠正这种输入错误,因而在关键期治疗弱视具有重要意义,重现关键期的可塑性一直以来是眼视觉科学研究的重点。
经颅直流电刺激被认为是治疗弱视有效的无创措施[49],在大鼠实验中发现该治疗方法能够增加PV+INs的数量且有效改善弱视眼视力。多次麻醉剂量氯胺酮在小鼠实验中被证实能够在不产生精神分裂等严重副作用下恢复视皮层可塑性,且停药后不影响PNNs再形成;给予视皮层60 Hz光刺激也能达到上述效果,这为无创干预弱视治疗提供了新思路[28]。PV+INs中条件性ErbB4敲除提示在成年期应用ErbB4抑制剂减少兴奋性突触,降低PV+INs兴奋性,可用以治疗成年弱视[39]。但有学者认为,抑制剂的应用仅仅只是提高可塑性,而移植胚胎MGE的中间神经元才能重新激活关键期,并在出生后 192 d 的小鼠中实现了视皮层关键期的激活,该过程与受体PV+INs上的ErbB4有关[50-51]。此外,体育活动也能提高视皮层可塑性,促进弱视大鼠视力的恢复,尽管PV+INs在该过程中活性没有变化,但生长因子的表达增强[52],表明外环境可塑性可能发生改变。
此外,Galuske等[53]认为,视觉皮层的可塑性是由神经元振荡局部控制的;而神经元振荡是由PV中间神经元的活动调节的,氯胺酮的应用和TrkB激活使包含gamma(γ)、theta (θ)、 alpha (α)和 beta (β)在内的神经振荡都有不同程度增强[41],成人视皮层关键期可塑性重启,但目前PV+INs产生的γ神经振荡与可塑性缺乏直接的联系。
5 结束语
PV+INs在皮层和海马区发挥重要作用,前人的研究普遍集中在海马区的认知障碍性疾病如阿尔茨海默病,近年来越来越多的文献肯定了其在视觉皮层发育中关键期可塑性的价值。鉴于特异性标志物PV与PV+INs膜固有特性密切相关,其在发育过程中参与视皮层关键期的机制鲜有研究;目前多是关注PV+INs外周环境如PNNs的改变。关于去除PNNs是否提高PV+INs的兴奋性仍存在争议,Balmer等[54]认为,PNNs提高了PV+INs的兴奋性;而在用硫酸软骨素去除PNNs的实验中观察到PV+INs兴奋性增强,明确了硫酸软骨素介导的可塑性不完全与突触可塑性相同。此外,皮层中仍有15%未包裹PNNs的PV+INs[22],它们在关键期可塑性调控中的作用仍待探索。尽管目前有关PV+INs治疗极少应用于临床,但我们仍要思考激活视觉皮层可塑性带来的问题:长期未关闭的关键期不利于双眼的图像整合[55],从而影响高级视觉功能如立体视觉,因此,激活关键期可塑性的最长时限是作为干预手段投入临床治疗弱视的一大挑战。
