基于16S rRNA 测序分析阻塞性睡眠呼吸暂停患者肠道靶标菌群的变化
2024-01-29朱继伟卢曼路焦倩倩孙运良丁红红
朱继伟,卢曼路,焦倩倩,孙运良,刘 璐,丁红红,于 燕,潘 磊
滨州医学院附属医院呼吸与危重症医学科,山东 滨州256603
阻塞性睡眠呼吸暂停(OSA)是一种常见的睡眠呼吸障碍类型,主要表现为睡眠时上呼吸道反复塌陷、气流停止,导致间歇性缺氧和睡眠碎片化[1,2]。研究表明,全球目前有近10亿人口患有OSA,其中我国患病人口最多[3]。OSA会导致氧化应激、炎症反应、代谢紊乱等多系统功能障碍[4,5],然而其机制目前尚不清楚。目前,临床针对OSA患者治疗方式大多数采用手术[6]或持续气道正压通气治疗[7],患者诊疗费用较高,依从性较差[8]。
近年来,随着高通量测序技术的发展,肠道菌群与OSA发生发展的关系也在逐渐被认识,OSA的发生发展与肠道菌群变化密切相关[9,10]。既往动物实验研究发现间歇性低氧小鼠模型肠道菌群出现明显变化,肠道菌群参与介导物质代谢和慢性炎症[11-13]。临床研究发现间歇性缺氧会改变肠道微生物组的生物节律[14],OSA患者肠道菌群中厚壁菌门丰度升高,拟杆菌门丰度降低,炎症相关细菌丰度明显升高[15-17]。Baldanzi 等[18]研究发现OSA患者肠道微生物物种多样性降低与呼吸暂停、缺氧密切相关。Ayyaswamy等[19]发现OSA诱导肠道微生态失调,导致出现神经免疫反应和肠道炎症。有研究表明OSA患者肠道菌群变化诱导肠道通透性增加,高血压合并OSA患者mTOR信号通路富集存在差异,与其病理生理变化密切相关[20]。目前涉及OSA患者的研究多为阐述OSA患者肠道菌群物种差异及与炎症关系,关于人肠道靶标菌属及其介导的相关差异代谢通路的研究尚未见报道。
本研究基于16S rRNA 高通量测序结果分析OSA患者及健康人群肠道菌群物种差异,基于多维度分析模型寻找验证OSA患者肠道靶标菌属,结合差异代谢通路分析标志菌群或肠道靶菌参与的代谢通路,以期OSA患者干预和治疗提供策略。
1 资料和方法
1.1 研究对象
随机选取2022年1~12月就诊于本院呼吸睡眠医学中心的39例OSA患者作为OSA组,同期检查无OSA的20例健康志愿者作为对照组。本研究通过滨州医学院附属医院伦理委员会批准(20220128-76),所有研究对象均知情同意。
1.2 纳入、排除标准
OSA 组纳入标准:经整夜多导睡眠监测初诊为OSA的患者,根据美国睡眠医学会睡眠及相关事件判读诊断标准,计算呼吸暂停低通气指数(AHI)为呼吸暂停和低通气的总发次数除以全部睡眠时间的总持续时间,AHI≥5次/h可诊断为OSA[21];未进行持续气道正压通气治疗。排除标准:年龄<18岁;服用影响睡眠的药物,如苯二氮䓬类药物、巴比妥类药物或镇静剂;接受相关治疗,包括但不限于手术、呼吸机治疗;合并胃肠道疾病,如溃疡性结肠炎和肠易激综合征等;入组前2月内服用肠道微生态制剂、抗生素或免疫抑制剂的患者;存在肿瘤、糖尿病、肝肾功能不全、风湿性免疫疾病或其他可能影响肠道菌群失调的疾病者。
对照组纳入标准:年龄>18岁;无睡眠呼吸暂停、失眠及其他睡眠障碍疾病病史。排除标准:入组前2 月内有服用微生态制剂、抗生素或免疫抑制剂者;入组前1月内有较大饮食结构或生活环境改变;存在消化系统疾病、肿瘤、糖尿病、肝肾功能不全、风湿性免疫疾病或其他可能影响肠道菌群失调的疾病者。
1.3 研究方法
1.3.1 样本采集与DNA提取 采集受试者清晨粪便样本0.5 g,储存于无菌无酶的粪便采样管中,避免尿液、月经等污染,标记好信息后,迅速转移至-80 ℃冰箱保存。所有粪便标本的冻融参照Wang等[22]的方法,采用Omega Mag-Bind Soil DNA Kit(200)(Omega)提取粪便样本中总微生物DNA,采用0.8%琼脂糖凝胶电泳分析样本DNA 相对分子质量和纯度,利用NanoDrop NC2000(Thermo Fisher Scientific)对DNA进行定量。
1.3.2 文库构建及基因测序 以微生物核糖体RNA等能够反映菌群组成和多样性的目标序列为靶点,应用16S rRNA V3-V4 区域特异引物进行聚合酶链式反应(PCR)扩增。PCR扩增选用细菌16S rDNA V3-V4区特异性引物,上游引物338F(5'-barcode +ACTCCTACGGGAGGCAGCA-3'),下游引物806R(5'-GGACTACHVGGGTWTCTAAT-3')。前引物中的barcode 是一个7 个碱基的寡核苷酸序列,用来区分同一文库中的不同样品。扩增结果进行2%琼脂糖凝胶电泳,然后用凝胶回收试剂盒(AXYGEN)回收目的片段。采用Illumina 公司的TruSeq Nano DNA LT Library Prep Kit建立测序文库。在Illumina MiSeq平台上利用MiSeq Reagent Kit V3(600cycles)对16S rRNAV3-V4区进行2×250 bp的双端测序。
1.4 生物信息学分析
1.4.1 肠道菌群测序质量评估 从每个样本中随机抽取一定数量的序列数据,绘制稀疏曲线,将各分组的分类操作单元(OUT)按其丰度从大到小沿横坐标依次排列后,将丰度值经Log10对数转换后的值作为纵坐标,在R软件中编写脚本绘制各样本或各分组的丰度等级曲线,反映测序数据量的合理性及丰富度,当曲线趋向平缓时,测序质量良好,测序数据量充足,可以用于后续分析。
1.4.2 物种组成分析 统计抽平后样本扩增子序列变异(ASV),获得每个样本的微生物群落的具体组成,对两组在门、属水平的物种进行可视化,使用circos图用来展示样本与物种的丰度关联关系。
1.4.3 Alpha多样性分析 使用QIIME 2进行Alpha多样性分析,以Shannon 指数和Simpson 指数表征多样性,以Chao1指数和Observed species指数表征菌群丰富度,以Pielou指数表征均匀度。
1.4.4 Beta多样性分析 通过主坐标分析(PCoA)、非度量多维尺度分析(NMDS)非约束排序手段对多维的微生物数据进行降维,展示数据变化的主要趋势。基于weighted UniFrac距离矩阵分析,通过二维排序图展示微生物群落的组成差异。
1.4.5 线性判别分析及影响因子(LEfSe)分析 LEfSe分析[23]对所有分类水平进行差异分析,识别组内组间物种丰度差异,来检测不同组间的差异丰富分类群,区分不同生物学特征的物种,寻找分组之间稳健的差异标志物种。
1.4.6 关联网络分析 采用ASV在本项目所有样本中的丰度数据,过滤掉序列总数少于10、出现样本数少于5 的ASV,采用SparCC算法,构建相关性矩阵,使用随机矩阵理论确定相关性数值的过滤阈值。使用subgraph函数过滤共现网络计算各样本子网络的图形水平拓扑学指数。根据Erdos Renyi模型以及当前网络的节点数目和边数目,构建随机网络。分别计算当前网络、随机网络中每个节点的相连节点数(度)。根据当前共现网络的模块化切割结果,计算当前网络中每个节点的Zi、Pi score值。根据Zi、Pi score值确定每个节点在关联网络中的角色[24]。
1.4.7 随机森林分析 使用未抽平的ASV,调用q2-sample-classifier中的“Classify_samples_ncv”函数进行随机森林分析以及巢式分层交叉检验[25]。进行10倍交叉检验,确定各物种在组间鉴别的重要性。
1.4.8 代谢通路预测分析 构建微生物基因组16S rRNA进化树,推测特征序列的最近序列物种,结合各样本特征序列丰度,计算基因家族拷贝数。将基因家族映射到MetaCyc数据库中,获得各样本中代谢通路的丰度数据。使用归一化的pathway丰度表,计算第二层次通路的平均丰度。运用MetagenomeSeq分析调用fitFeatureModel 函数使用zero-inflated log-normal model 对每个pathway的分布进行拟合,并使用该模型的拟合结果判别差异的显著性。依据选择的通路抓取分层的样本代谢通路丰度表中的相应数据,绘制物种组成柱状图。
1.5 统计学分析
采用SPSS26.0软件对数据进行统计学分析。用QIIME 2 和R(3.2.0)软件进行序列数据分析。符合正态分布的计量资料采用均数±标准差表示,两组间比较采用独立样本t检验;非正态分布资料应用非参数检验分析,两组间比较采用χ2检验。以Ρ<0.05为差异具有统计学意义。
2 结果
2.1 临床资料比较
本研究最终获得59例有效样本,其中OSA组样本39例,对照组样本20例,两组人群性别、年龄差异均无统计学意义(Ρ>0.05),与对照组相比,OSA组BMI、AHI指数明显升高,夜间平均血氧降低(Ρ<0.01,表1)。

表1 OSA组和健康对照组一般资料Tab.1 General data of OSAgroup and healthy control group(Mean±SD)
2.2 肠道菌群测序质量评估
两组样本的稀释曲线及丰度等级曲线均趋向平坦,说明物种丰富度足够,测序样本量及数据量渐进合理(图1)。肠道菌群测序质量良好,测序数据量充足,可以用于后续分析。
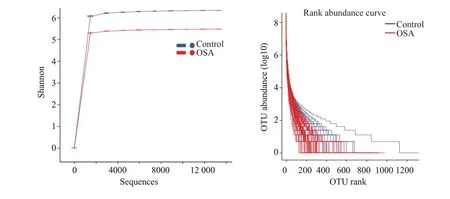
图1 稀释曲线及丰度等级曲线Fig.1 Rarefaction curve and RankAbundance curves.OUT:Operational taxonomic unit.
2.3 物种组成分析
对受试者样本进行16S rRNA 基因测序,得到有效序列总数为12 615 425。在门水平上,测序结果显示两组菌群主要是由厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形菌门(Ρroteobacteria)、放线菌门(Actinobacteria)4类菌群组成,与对照组相比,OSA组Firmicutes、Ρroteobacteria、Actinobacteria丰度比例升高,而Bacteroidetes丰度比例降低(图2A)。在属水平上,样本菌群测序结果相对丰度前10的菌群依次为拟杆菌属(Bacteroides)、粪杆菌属(Faecalibacterium)、罗斯氏菌属(Roseburia)、双歧杆菌属(Bifidobacterium)、志贺氏菌属(Shigella)、布劳特氏菌属(Blautia)、巨单胞菌属(Megamonas)、瘤胃球菌属(Ruminococcus)、普雷沃氏菌属(Ρrevotella)、戴阿里斯特杆菌属(Dialister),与对照组相比,OSA组Faecalibacterium、Roseburia丰度明显降低,Megamonas丰度明显升高(图2B)。
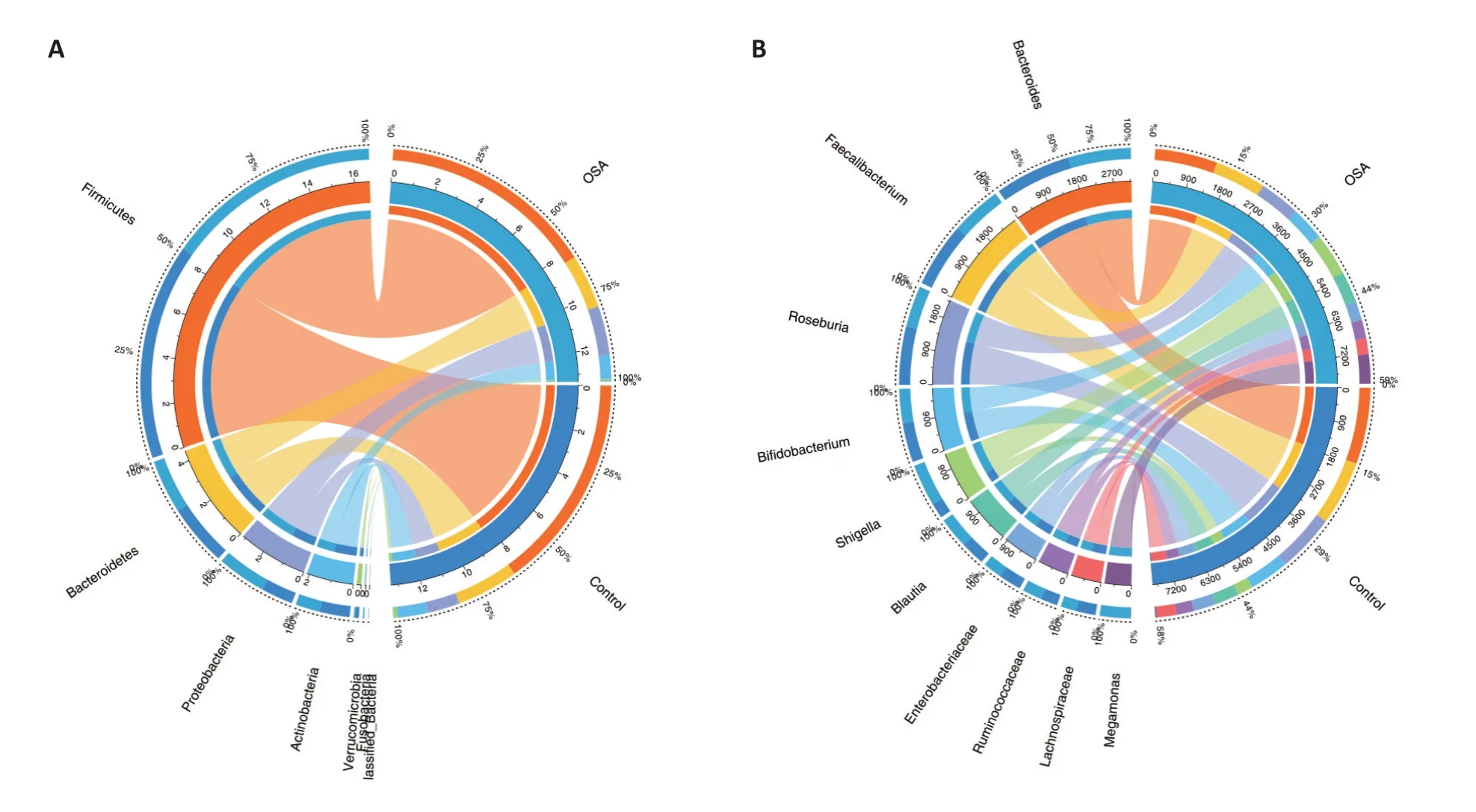
图2 OSA组与对照组物种组成分布Fig.2 Composition and distribution of TOP10 species in terms of relative abundance in OSA group and control group.A:Phylum;B:Genus.
2.4 肠道微生物Alpha多样性分析
Alpha多样性分析结果显示,OSA组物种多样性Shannon 和Simpson 指数低于健康对照组(Ρ<0.01);OSA组物种丰富度Chao1 和Observed species低于健康对照组,Observed species两组间差异具有统计学意义(Ρ<0.05)。OSA组菌群均匀度低于对照组(Ρ<0.05,表2)。
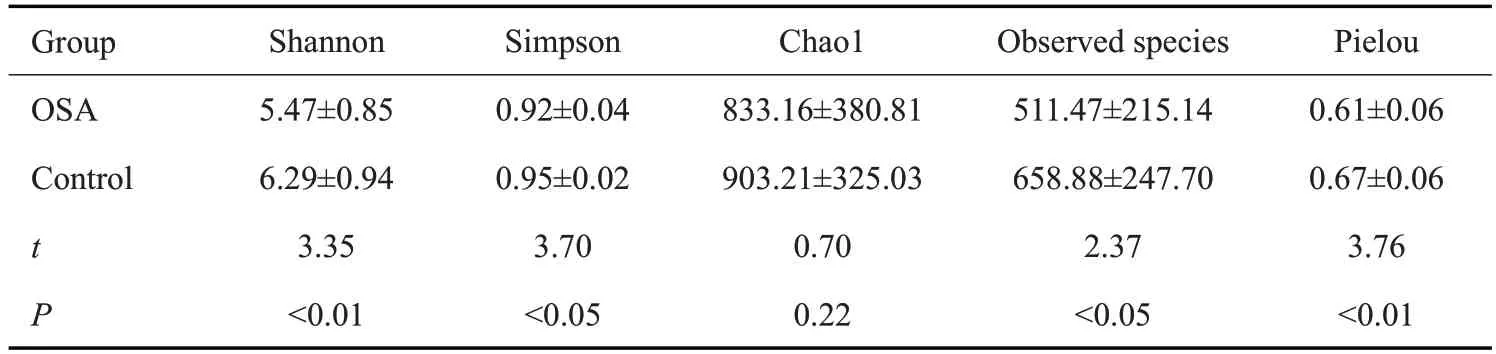
表2 OSA组和对照组的Alpha多样性分析Tab.2 Alpha diversity analysis in OSAgroup and control group(Mean±SD)
2.5 肠道微生物Beta多样性分析
PCoA分析结果显示,OSA组与对照组相距较远,两样本差异较大(Ρ<0.05,图3A);NMDS分析应力值Stress=0.118(图3B),当应力值<0.2时差异分析结果可靠,具有统计学意义(Ρ<0.05)。
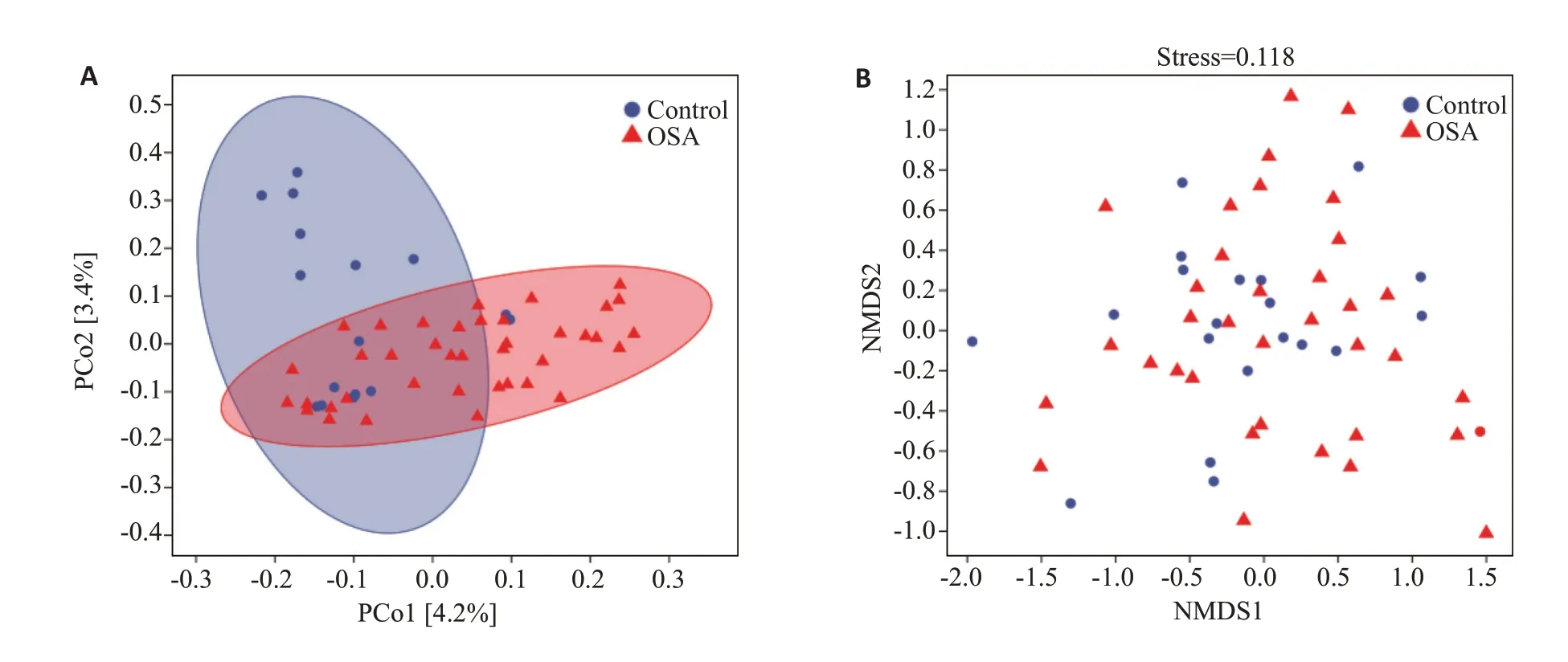
图3 OSA组和对照组的Beta多样性分析Fig.3 Beta diversity analysis in OSA group and control group.A:Principal co-ordinate analysis;B:Nonmetric multidimensional scaling(NMDS).
2.6 物种差异与标志物种分析
2.6.1 组间差异分析 Anosim分析显示,两组肠道菌群组间差异大于组内差异,差异有统计学意义(R=0.268,Ρ=0.001,图4)。
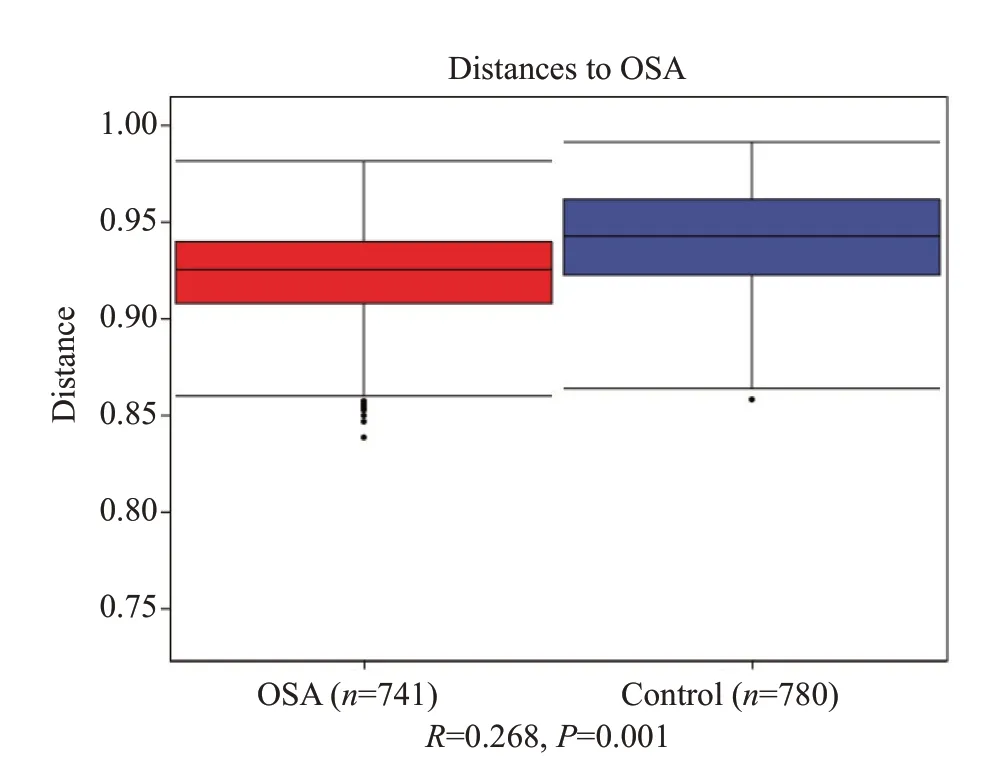
图4 OSA组和对照组的Anosim分析Fig.4 Anosim analysis in OSAgroup and control group.
2.6.2 物种差异分析 在门水平上,Firmicutes、Ρroteobacteria、梭杆菌门(Fusobacteria)、TM7、酸杆菌门差异有统计学意义(Ρ<0.05,图5A)。在属水平上,OSA组和健康对照组在26个菌属中的差异有统计学意义(Ρ<0.05),丰度较高的前7位菌属中,OSA组假单胞菌属(Ρseudomonas)、Megamonas、Ruminococcus、毛螺菌科(属)(Lachnospiraceae)、多尔氏菌属、丁酸球菌属高于对照组(Ρ<0.05,图5B)。
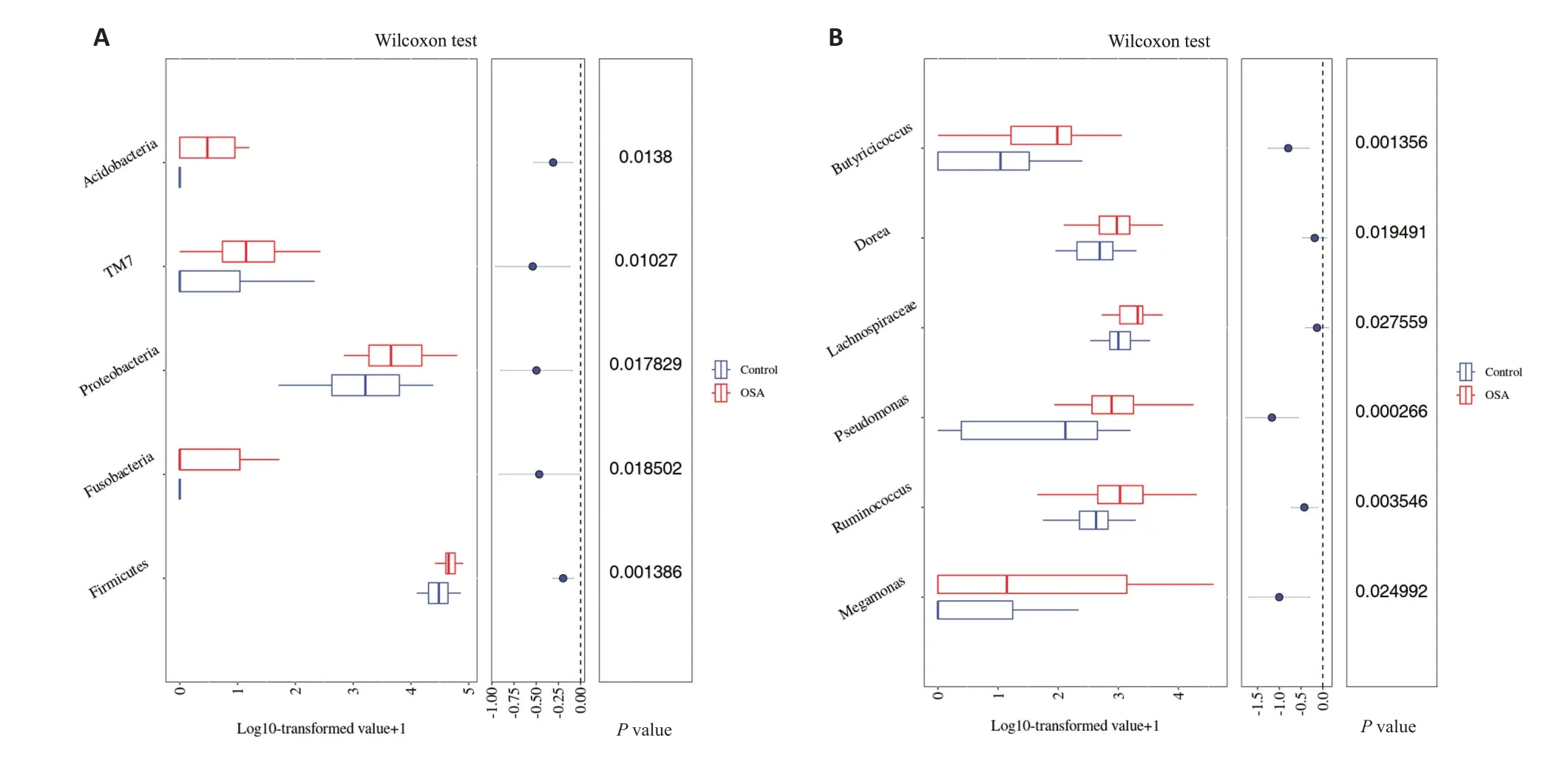
图5 组间物种差异分析Fig.5 Species difference analysis at the genus level.A:Phylum;B:Genus.
2.6.3 LEfSe分析 LEfSe分析柱状图及进化分支图结果显示,两组肠道菌群中23种菌群丰度比较,差异有统计学意义(Ρ<0.05)。其中OSA 组韦荣氏球菌科(Alicyclobacillaceae)、Megamonas、假单胞菌科(Ρseudomonadaceae)、假单胞菌目(Ρseudomonadales)、Ρseudomonas、肠球菌科(Enterococcaceae)、肠球菌属(Enterococcus)、梭杆菌纲(Fusobacteriia)、梭杆菌目(Fusobacteriales)、梭杆菌门(Fusobacteria)、梭杆菌属(Fusobacterium)等17 种肠道菌群丰度高于对照组(Ρ<0.05);对照组栖粪杆菌属(Faecalibacterium)、杆孢囊菌属(Virgisporangium)、RF3、1_68、脂环酸芽孢杆菌属(Alicyclobacillus)、脂环酸芽孢杆菌科(Alicyclobacillaceae)肠道菌群丰度高于OSA 组(Ρ<0.05,图6)。
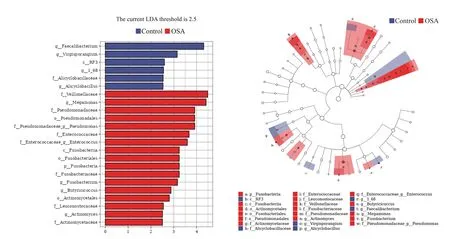
图6 两组人群肠道菌群LEfSe 分析柱状图及进化分支图Fig.6 LEfSe analysis histogram and evolutionary branching diagram of gut microbiota in the two groups.LDA:Linear discriminant analysis.
2.6.4 关联网络分析 构建相关性矩阵确定阈值为0.565(图7A),拓扑指数分析显示,肠道菌群度分布结果符合无标度网络,说明影响宿主稳态的主要因素为差异性标志菌群(图7B)。ZiPi图结果显示,在门水平上厚壁菌门作为主要影响宿主稳态的关键菌群(图7C),在属水平上影响宿主稳态的关键物种为栖粪杆菌属、志贺菌属、假单胞菌属(图7D)。

图7 关联网络分析Fig.7 Analysis of the association networks.A: Correlation matrix threshold;B: Topological exponential analysis;C:ZiPi;D:Key species that affect the host homeostasis.
2.6.5 差异标记物种分析 随机森林模型分析结果显示Pseudomonas在鉴别两组中的重要性最高(图8A)。在属水平上对LEfSe分析差异物种进行受试者工作特征(ROC)曲线分析,根据曲线下面积评估差异性菌群诊断OSA的敏感性。其中Pseudomonas AUC=0.798[95%CI(0.674-0.921),Ρ=0.0003]、Megamonas AUC=0.678[95%CI(0.537-0.818),Ρ=0.029],可作为重要鉴别意义的生物标记物(图8B)。
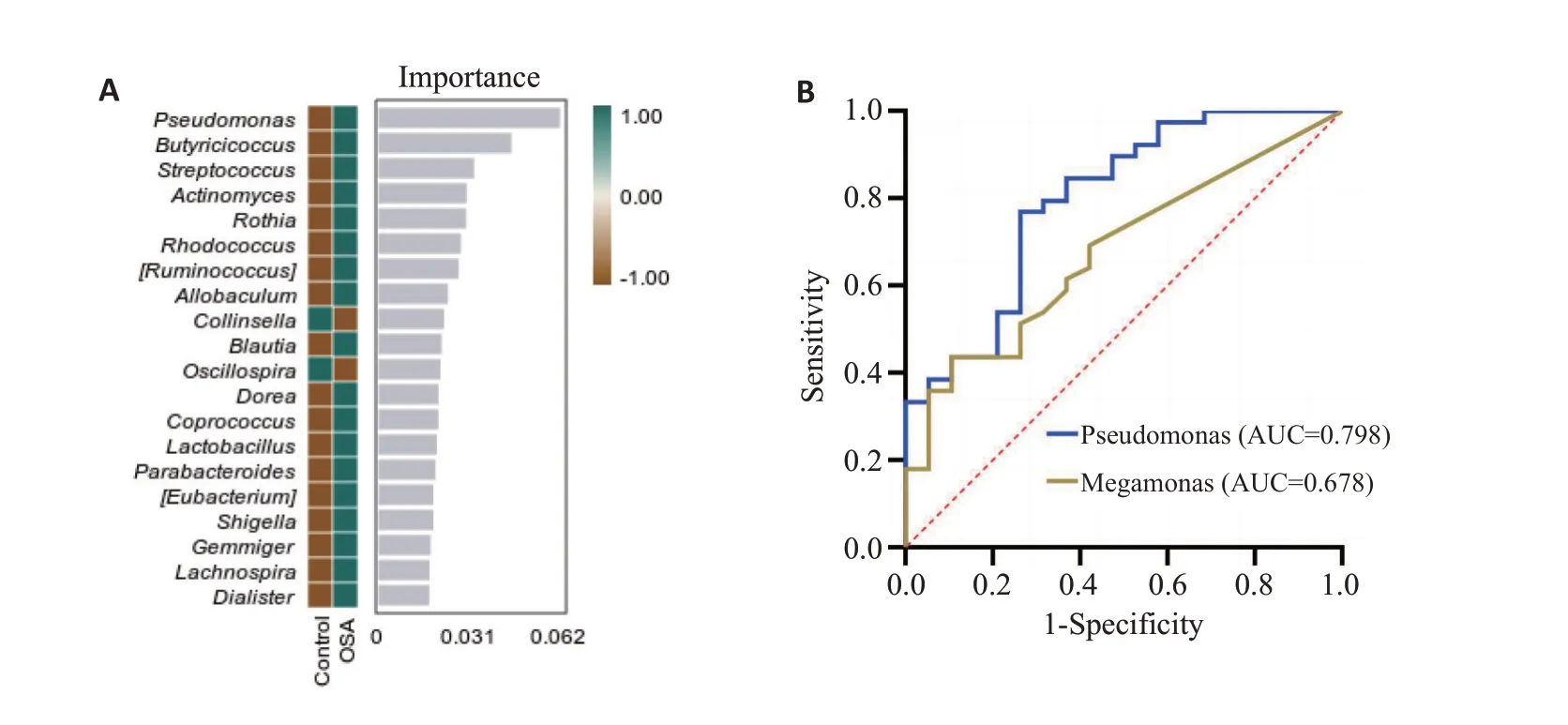
图8 差异标记物种分析Fig.8 Analysis of the differentially labeled species.A:Random forest model;B:ROC analysis.
2.7 差异生物功能代谢通路功能预测分析
基于MetaCyc数据库,通过PICRUSt2预测分析显示,在宿主体内肠道菌群稳态起主要作用的肠道菌群的功能是生物合成功能,起主要作用的为氨基酸生物合成、核苷酸生物合成等。其他预测功能包括降解、利用、同化、解毒、前体代谢产物和能量的产生、聚糖途径、大分子修饰、代谢簇(图9A)。MetagenomeSeq分析结果显示,在芳香族生物胺降解、酮葡萄糖酸代谢(Ρ<0.001),葡萄糖降解、没食子酸酯降解、L-精氨酸和L-鸟氨酸降解超途径、L-酪氨酸降解I、腺苷钴胺生物合成II、L-亮氨酸降解I、甲苯降解IV(Ρ<0.05,图9B)。物种组成结果显示Pseudomonas可能是影响物质通路代谢的关键菌属(图9C、D)。
3 讨论
本研究基于16S rRNA高通量测序,分析OSA患者与健康对照组肠道菌群物种差异,发现两组人群肠道菌群在组成结构和物种多样性方面存在显著差异。OSA组潜在致病菌属(如假单胞菌属、巨单胞菌属)丰度增加,多维度分析模型及差异代谢通路分析发现假单胞菌属可能通过参与芳香族生物胺降解和酮葡萄糖酸代谢通路影响OSA的发生发展,可望作为OSA患者肠道菌群的潜在靶标。
基于Shannon稀释曲线及丰度等级曲线分析结果表明本研究测序数据量完整,测序质量良好,研究结果可靠。α多样性分析结果显示OSA组物种多样性、丰富度、均匀度均低于健康对照组,β多样性分析和Anosim分析结果表明两组间菌群结构存在差异且组间差异大于组内差异。物种组成差异分析结果显示,OSA组厚壁菌门、梭杆菌门、假单胞菌属、瘤胃肠球菌属等丰度明显升高,产短链脂肪酸丰度明显降低。这与Collado等[26]研究结果相似,研究发现厚壁菌门/拟杆菌门比例明显升高,OSA患者肠道微生物群物种丰富度明显低于对照组。有研究同样证明OSA儿童患者肠道菌群组成发生明显改变,产短链脂肪酸菌属丰度降低[27]。结合既往研究,表明OSA患者肠道菌群稳态发生改变,菌群微生态紊乱失衡。
为进一步确定影响OSA患者肠道菌群稳态的靶标菌属,本研究运用多维度模型分析寻找验证OSA患者肠道菌群生物标记物。LEfSe分析结果表明在属水平上OSA组假单胞菌属、巨单胞菌属丰度显著升高。关联网络分析结果显示,影响宿主稳态的关键物种为栖粪杆菌属、志贺菌属、假单胞菌属等。同时随机森林模型分析机器学习算法及ROC曲线分析结果同样显示假单胞菌属在组间差异菌属中重要性和敏感度最高。这与Li等[28]研究结果相似,在OSA患者肠道菌群中同样假单胞菌属丰度升高,假单胞菌属丰度和D-乳酸呈正相关,D-乳酸是组织缺氧的敏感指标,间接证明假单胞菌属和组织缺氧密切相关。Ren等[29]研究发现假单胞菌属与肠道屏障功能障碍和感染有关,可能导致败血症和多器官功能障碍综合征。需要注意的是,有研究发现瘤胃球菌属同样与肠道屏障功能密切相关,本研究OSA组瘤胃球菌属丰度明显升高[30]。Wen等[31]研究发现假单胞菌属感染会引起机体的Th17细胞反应和系统性炎症反应。同样有研究发现假单胞菌属与炎症性肠病发病密切相关,通过影响肠道菌群的稳态来导致肠道免疫系统出现异常反应[32]。有研究发现假单胞菌属参与肠-肺轴免疫调节,影响呼吸系统疾病的发生发展[33]。结合上述研究说明假单胞菌群可能通过介导免疫炎症反应,参与肠-肺轴免疫调节导致OSA患者的肠道屏障功能受损和代谢紊乱。
在差异生物功能代谢通路功能预测分析中,二级功能通路分析显示在生物合成途径的相对丰度最高,占比最多的是氨基酸生物合成。组间显著差异通路分析结果显示OSA组芳香族生物胺降解、酮葡萄糖酸代谢通路显著上调。分层样本代谢通路分析结果发现在这两个通路中起主要作用的均为假单胞菌属。研究发现芳香族生物胺是生物体内活性成分,在物质合成及生物膜稳定性方面起重要作用,具有抗氧化及调节肠道生理功能的作用[34-36]。因此,OSA患者芳香族生物胺降解通路丰度升高,芳香族生物胺降解增多,影响OSA患者物质合成、物质代谢及抗氧化能力。Sun等[37]研究发现假单胞菌属与酮葡萄糖代谢密切相关,在缺氧条件下假单胞菌属、巨单胞菌属通过酮葡萄糖酸代谢通路氧化生成2-酮基-D-葡萄糖酸,利用2-酮基-D-葡萄糖酸对细胞生长及代谢产生负面影响。因此,推测假单胞菌属可能通过上述代谢通路,介导OSA患者的病理生理学改变,有助于解释肠道菌群与OSA患者的内在关系,有望为干预OSA提供新的策略。
综上所述,本研究发现与健康人群相比,OSA患者肠道菌群的整体结构失衡,物种组成紊乱且有差异,考虑差异与OSA的病理生理如间歇性缺氧及睡眠碎片化等因素有关;假单胞菌属可作为重要鉴别意义的生物标记物,作为肠道靶标菌群可能参与芳香族生物胺降解和酮葡萄糖酸代谢通路,进而影响OSA的发生发展。另外,本研究存在一定局限性:首先,两组存在性别差距,OSA组男性患者较多,这也与OSA的发病人群有关[38];其次,两组年龄和BMI有一定差异,可能影响两组肠道菌群物种组成,后续研究将进一步探讨年龄和BMI对OSA肠道菌群的影响。最后,肠道菌群容易受到环境、遗传、饮食行为习惯等因素的影响,本研究地域单一,且受限于方法学,本研究所取样品可能无法反映肠道内厌氧菌状态。然而有关OSA肠道菌群研究方兴未艾,尤其专注肠道菌群代谢通路研究,将为OSA的早期干预和治疗提供新思路。
