六味能消制剂中非法成分土大黄苷检测方法的建立
2024-01-27祁晓玲杨凤梅
祁晓玲,张 炜,杨凤梅
青海省药品检验检测院,青海省中藏药现代化研究重点实验室,国家药品监督管理局中药(藏药)质量控制重点实验室,西宁810016
六味能消制剂分为丸剂、胶囊剂、片剂3 种剂型。其处方由藏木香、干姜、诃子、大黄、寒水石、碱花6 味药材组成。其主要功效为助消化、消肿、理气和胃,用于食物中毒症、积食不化、胃痛、胸腹肿胀、大便干燥的治疗,还可用于高脂血症及肥胖症的治疗。大黄为该处方中重要的药味之一,收载于《中华人民共和国药典》(以下简称《中国药典》)2020年版一部,为蓼科植物掌叶大黄Rheum palmatumL.、唐古特大黄Rheum tanguticumMaxim. ex Balf. 或药用大黄Rheum officinaleBaill. 的干燥根和根茎;具有泻下攻积、清热泻火、凉血解毒、逐瘀通经、利湿退黄的功效[1]。近年来,大黄药材资源无法满足日益增长的市场需求,使得市场中出现用同属不同种其他植物的根部冒充大黄的现象,存在非药用大黄代替大黄投料的情况,如藏边大黄、河套大黄、华北大黄、天山大黄、信州大黄、土大黄等,这些品种《中国药典》未收藏,不可作为大黄药用。伪品大黄来源广、价格低,虽与大黄为同科植物,但其含土大黄苷,结构为二苯乙烯苷类,几乎无泻下作用,甚至会引发不良反应,故不可代替大黄药用[2]。为了全面评价六味能消丸(胶囊、片)的产品质量和现行标准的可行性,警示六味能消丸(胶囊、片)存在的安全风险,本研究建立了用薄层色谱法( thin layer chromatography,TLC)、高效液相色谱(high performance liquid chromatography,HPLC)对六味能消制剂中土大黄苷成分进行初筛及用高效液相色谱法-串联质谱法(HPLC-MS)对检出土大黄苷的阳性样品进行进一步确证的方法[3-7]。
1 仪器与试药
1.1 仪器
Ultimate3000-TSQ Quantum Ultra 型液质联用仪(三重四极杆,美国THERMO 公司);岛津LC-20ATVP 型高效液相色谱仪(二极管阵列检测器,日本岛津公司);SQP 型百分之一电子天平、SQP 型万分之一电子天平、MSA225-1CE-DI 型十万分之一电子天平(德国Sartrious 公司);DT-1028-CH 型超声波提取器( 德国 BANDELIN 公司);LINOMAT 5 型半自动点样仪、TLC VISUALIZER薄层数码成像仪(瑞士卡玛公司);硅胶G 板(德国默克公司)。
1.2 试药
土大黄苷对照品(批号110794-201909,中国食品药品检定研究院,供检查用,批号R05J9F65074,HPLC 测定质量分数≥98%,上海源叶生物科技有限公司);甲醇、乙腈为色谱纯,均购自德国默克公司;水为超纯水(用Milli-Q 超纯水机自制);其他试剂均为分析纯。六味能消丸(胶囊、片)均为市售样品,涉及4 家企业,共101 批次。
2 方法与结果
2.1 TLC 法初步筛查
2.1.1 对照品溶液的制备 取土大黄苷对照品适量,加甲醇制成每1 mL 含0.2 mg 土大黄苷的溶液,作为对照品溶液。
2.1.2 供试品溶液的制备 取本品适量,研细,取1 g,精密称定,置于具塞锥形瓶中,精密加入甲醇50 mL,密塞,称定质量,超声处理(功率为300 W,频率为35 kHz)40 min,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过,取滤液5 mL 备用,其余滤液挥干,残渣加甲醇1 mL 使溶解,作为供试品溶液。
2.1.3 对照药材溶液的制备 取大黄对照药材0.5 g,按照2.1.2 项下供试品溶液制备方法制成对照药材溶液。
2.1.4 阴性对照溶液的制备 取原料药材,按处方制得缺大黄的六味能消丸(胶囊、片)阴性样品。取1 g,按照2.1.2 项下供试品溶液制备方法制成大黄阴性溶液。
2.1.5 模拟制剂溶液的制备 取原料药材,按处方、工艺制得六味能消丸(胶囊、片)模拟制剂样品。取1 g,按照2.1.2 项下供试品溶液制备方法制成模拟制剂溶液。
2.1.6 色谱条件及结果 吸取上述制备的供试品溶液、阴性对照溶液、模拟制剂溶液、大黄药材溶液、土大黄苷对照品溶液各10 μL,分别点于同一硅胶G 薄层板上,以二氯甲烷-甲醇-甲酸-水(10∶3.5∶0.2∶0.3)为展开剂,展开,取出,晾干,置于紫外光灯365 nm 处检视。用以上方法检验发现,A 企业生产的共13 个批次六味能消丸中有12 个批次样品在与土大黄苷对照品色谱相应位置上检出相应的斑点。其余3 个企业的六味能消丸(胶囊、片)中均未检出土大黄苷,且阴性样品无干扰。见图1。

图1 六味能消制剂薄层色谱图-365 nm(默克板,26 ℃,40%)Fig. 1 TLC chromatograms of Liuwei Nengxiao preparationsunder 365 nm(Merck,26 ℃,40%)
2.1.7 TLC 考察
2.1.7.1 展开剂的选择 本实验共考察了2 种展开剂,展开剂1 为甲苯-甲酸乙酯-丙酮-甲醇-甲酸(30∶5∶5∶20∶0.1),展开剂2 为二氯甲烷-甲醇-甲酸-水(10∶3.5∶0.2∶0.3),结果用展开剂1 斑点分离较差,相较而言,用第2 种展开剂斑点分离较好,且阴性样品在与土大黄苷对照品相应的位置上无干扰,见图2。故将展开剂2 作为本方法的展开剂。

图2 展开剂的比较(26 ℃,40%)Fig.2 Comparison of developing solvents(26 ℃,40%)
2.1.7.2 点样量的选择 吸取土大黄苷对照品溶液(0.2 mg·mL-1),分别点样1、2、5、8、10 μL,条带宽度约为8 mm;吸取供试品溶液(批号2020030101),分别点样1、2、5、8、10 μL,条带宽度约为8 mm,结果表明,对照品和供试品点样量均为10 μL,薄层斑点清晰,分离效果较好。见图3。

图3 供试品薄层色谱图-365 nm(默克板,26 ℃,40%)Fig.3 TLC chromatograms of samples-under 365 nm(Merck,26 ℃,40%)
2.1.7.3 耐用性考察 分别用青岛海洋化工厂、德国默克公司的硅胶G 薄层板进行实验,结果显示,不同厂家生产的硅胶G 薄层板斑点均显现清晰。见图4。

图4 六味能消制剂薄层色谱图-365 nm(青岛海洋板,24 ℃,35%)Fig.4 TLC chromatograms of Liuwei Nengxiao preparations-under 365 nm (plate: Qingdao Ocean,24 ℃,35%)
2.1.7.4 检测限 吸取土大黄苷对照品溶液(0.2 mg·mL-1),分别点样1、2、5、8、10 μL(即0.2、0.4、1.0、1.6、2.0 μg),条带宽度约为8 mm;吸取供试品溶液(批号2020030101),分别点样1、2、5、8、10 μL(即0.1、0.2、0.5、0.8、1.0 mg),按照2.1.6 项下条件检视,以人眼可检视到的最低斑点点样量计[8],得到土大黄苷的检测限(limit of detection, LOD)为0.2 μg,见图5。但由于人眼对于不同颜色的辨识存在一定差异,不同操作人员可能得出不同的结果,故在实际操作中,仍需用HPLC-MS 进行进一步确证。
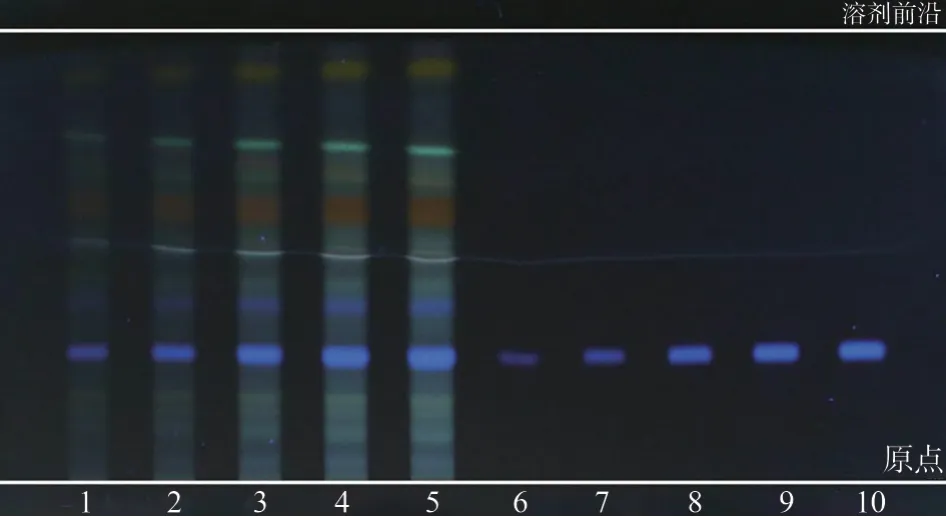
图5 LOD 薄层色谱图-365 nm(默克板,26 ℃,40%)Fig.5 TLC chromatograms of LOD-under 365 nm(Merck,26 ℃,40%)
2.2 HPLC 法初步确认
2.2.1 色谱条件 十八烷基硅烷键合硅胶为填充剂的色谱柱Agilent Extend-C18(5 μm,250 mm×4.6 mm);流动相为甲醇-水(30∶70);流速为1.0 mL·min-1;柱温为25 ℃;进样量为10 μL;检测波长为324 nm(将配制好的土大黄苷对照品溶液进行吸收波长扫描,结果土大黄苷在(324±2)nm 波长处有最大吸收,故选择324 nm 为检测波长)。见图6。

图6 土大黄苷的紫外吸收图Fig.6 UV absorption spectrum of rhaponticin
2.2.2 对照品溶液的制备 精密称取土大黄苷对照品18.14 mg,置于100 mL 量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得储备液。精密量取储备液3 mL,置于50 mL 量瓶中,用甲醇稀释至刻度,摇匀即得(每1 mL 约含土大黄苷10.66 μg)。
2.2.3 供试品溶液的制备 取2.1.2 项下供试品溶液的备用滤液,即得。
2.2.4 大黄药材溶液的制备 取大黄对照药材、各厂家提供的大黄1 g,按照2.1.3 项下对照药材溶液的制备方法制备,即得。
2.2.5 阴性对照溶液的制备 取2.1.4 项下阴性对照溶液的备用滤液,即得。
2.2.6 模拟制剂溶液的制备 取2.1.5 项下模拟制剂溶液的备用滤液,即得。
2.2.7 提取溶剂的选择 分别考察了2 种常用的提取溶剂甲醇、乙醇,取供试品(批号2020030101),按照2.1.2 项下溶液制备方法制备,注入高效液相色谱仪,结果显示,用甲醇提取的供试品色谱峰峰面积为4 021 589,理论塔板数为8 474;用乙醇提取的供试品峰面积为1 845 996,理论塔板数为4 480,用甲醇提取的供试品色谱峰峰面积和理论塔板数均较高,故选择甲醇作为提取溶剂。
2.2.8 专属性考察 按照2.2.1 项下色谱条件,分别精密吸取上述溶液各10 μL,注入液相色谱仪,测得阳性样品中呈现与土大黄苷对照品保留时间一致的色谱峰,且阴性无干扰。见图7 至图10。再用二极管阵列检测比较相应色谱峰在200~400 nm 波长范围内的紫外-可见吸收光谱,测得土大黄苷对照品与阳性样品吸收光谱在(324±2) nm 处均有最大吸收,由此可判断二者为同一物质。见图6、图11。

图7 土大黄苷对照品的HPLC 图Fig.7 HPLC chromatogram of rhaponticin reference
图8 大黄对照药材的HPLC 图Fig.8 HPLC chromatogram of Rheum palmatum

图9 阴性样品HPLC 图Fig.9 HPLC chromatogram of negative sample
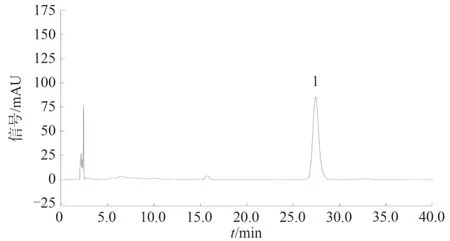
图10 阳性样品的HPLC 图Fig.10 HPLC chromatogram of positive sample
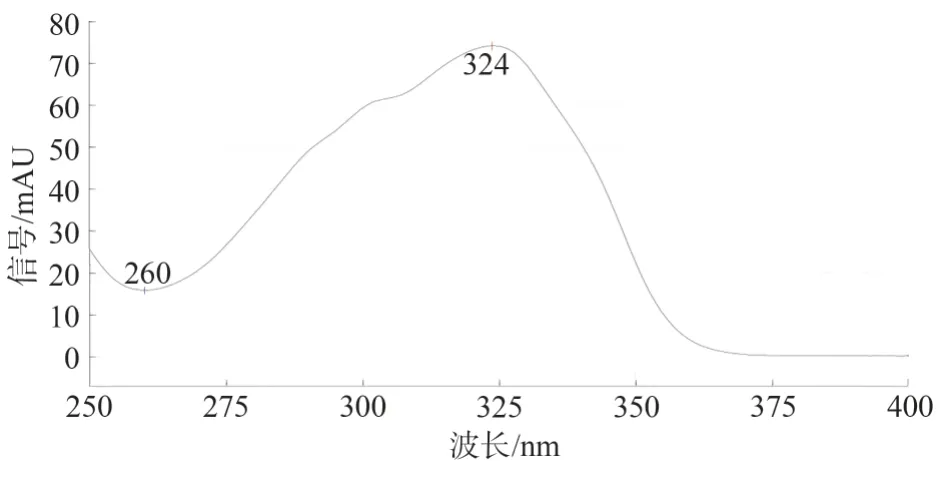
图11 阳性样品的紫外吸收图Fig.11 UV absorption spectrum of positive sample
2.2.9 线性考察 精密吸取不同质量浓度的土大黄苷对照品溶液(2.13、4.26、8.53、12.79、17.06、21.32 μg·mL-1)10 μL,分别在2.2.1项下色谱条件下测定,以土大黄苷对照品质量浓度为横坐标(x,μg·mL-1),以峰面积为纵坐标(y)进行线性回归,土大黄苷在2.13~21.32 μg·mL-1范围内回归方程为y=39 904x-747.42(r=0.999 9),表明线性关系良好。
2.2.10 LOD 取2.2.2 项下的土大黄苷对照品储备液,逐步稀释成85.33 ng·mL-1的对照品溶液。吸取上述溶液10 μL,注入液相色谱仪,计算得信噪比为3∶1,确定85.33 ng·mL-1为仪器检出限,故方法LOD 为4.266 μg·g-1。
2.2.11 精密度实验 精密吸取2.2.2 项下的土大黄苷对照品(质量浓度约为10.66 μg·mL-1),按照2.2.1 项下色谱条件连续进样5 次,测得土大黄苷保留时间的RSD 值为0.29%,峰面积的RSD 值为0.47%,结果表明精密度良好。
2.2.12 稳定性实验 取同一份供试品溶液,分别于0、2、4、8、12、24 h,按照2.2.1 项下色谱条件进样测定峰面积,测得土大黄苷峰面积的RSD 值为0.31%,表明供试品中土大黄苷在24 h内稳定性良好。
2.2.13 重复性实验 取同一份供试品,精密称取6 份,按照2.1.2 项下方法制成供试品溶液,按照2.2.1项下色谱条件测定,测得土大黄苷含量平均值为0.039%,RSD 值为0.16%,表明实验的重复性良好。
2.2.14 加样回收实验 取阴性样品1.0 g,精密称定,共6 份,分别精密加入质量浓度为26.08 μg·mL-1的土大黄苷对照品溶液50 mL,超声处理(功率为300 W,频率为35 kHz)40 min,放冷,再称定质量,用甲醇补足减失的质量,摇匀,用微孔滤膜(0.45 μm)滤过,取续滤液作为供试品溶液,进样10 μL,测定,计算回收率,结果表明本方法的准确度良好。见表1。

表1 土大黄苷的加样回收率实验结果 (n=6)Tab.1 Results of rhaponticin recovery test (n=6)

表2 六味能消制剂中土大黄苷的检查结果Tab.2 Results of rhaponticin in Liuwei Nengxiao preparations
2.2.15 耐用性实验 由于不同生产厂家的高效液相色谱仪和色谱柱存在性能上的差异,为考察方法的可行性及通用性,对供试品还进行了耐用性实验:①用不同厂家生产的色谱柱;②在不同的仪器上测定。结果表明耐用性较好。见图12、图13。图12、图13 分别为Waters e2695-2998 型高效液相色谱仪、Waters X-Bridge C18(250 mm×4.6 mm,5 μm)色谱柱所得色谱图。
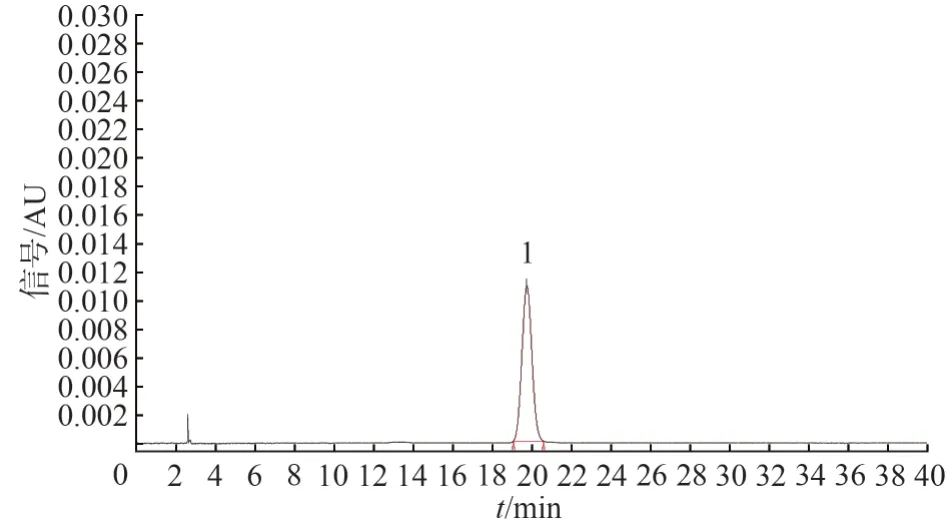
图12 土大黄苷对照品的HPLC 图Fig.12 HPLC chromatogram of rhaponticin reference
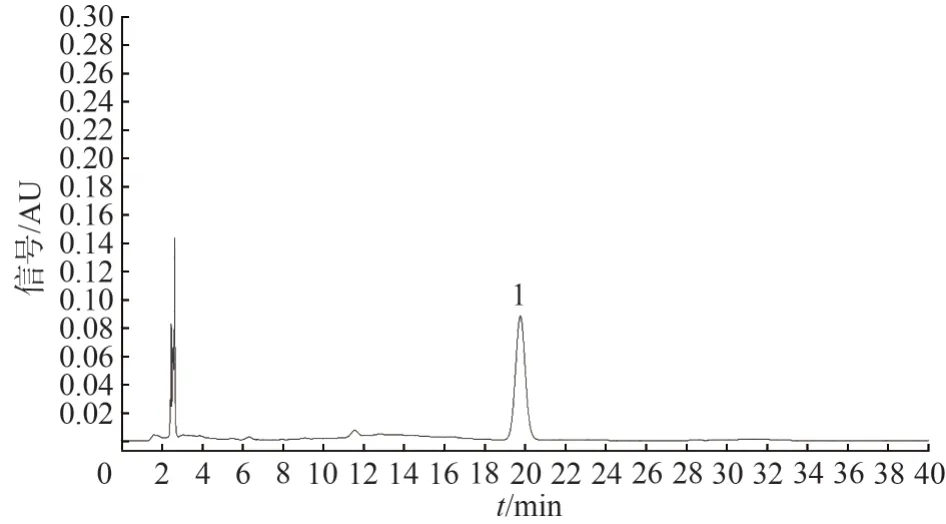
图13 阳性样品的HPLC 图Fig.13 HPLC chromatogram of positive sample
2.2.16 样品测定 取六味能消丸(胶囊、片)样品适量,按照2.1.2 项下方法制成供试品溶液,用2.2.1项下色谱条件测定,结果在4 家企业共101 批次样品中,A 企业生产的12 个批次样品中检出土大黄苷色谱峰,且紫外吸收光谱一致,呈阳性检出,该结果与TLC 检测结果一致。其余3 个企业样品中均未检出土大黄苷色谱峰。
2.3 HPLC-MS 方法验证
2.3.1 色谱条件
Thermo Hypersil GOLD AQ C18(100 mm×2.1 mm,1.9 μm);以乙腈(A)-1 mL·L-1甲酸溶液(B)为流动相,梯度洗脱(0~6 min,20% A~44% A);流速为0.3 mL·min-1;柱温为35 ℃。
2.3.2 质谱条件
电喷雾离子源(electrospray Ionization,ESI),离子源温度为110 ℃;毛细管温度为250 ℃;鞘气流速为20 L·min-1;辅助气流速为10 L·min-1;喷雾电压为3 500 V;锥孔电压为40 V;碰撞能量(collision energy,CE)为30 V;正离子扫描模式,扫描模式分为Full Scan(一级全扫描)和Product Scan(二级全扫描);一级全扫描质量范围m/z400~500,二级全扫描质量范围m/z100~300。
2.3.3 对照品溶液的制备
同2.2.2 项下对照品溶液制备方法。
2.3.4 供试品溶液的制备
取2.1 项下TLC 和2.2 项下HPLC 法检出土大黄苷的供试品溶液,即得。
2.3.5 阳性对照溶液的制备
取2.1 项下TLC 和2.2 项下HPLC 法未检出土大黄苷的样品1 g,加入0.2 mg·mL-1土大黄苷甲醇溶液2.5 mL,同2.1.2 项下供试品溶液的制备方法,取滤液,即得。
2.3.6 阴性对照溶液的制备
取2.1 项下TLC 和2.2 项下HPLC 法未检出土大黄苷的供试品溶液,即得。
2.3.7 空白试剂溶液的制备
取甲醇作为空白试剂溶液。
2.3.8 测定条件
按照上述色谱及质谱条件,分别精密吸取上述溶液各5 μL,注入液质联用仪,测定,即得。供试品溶液一级全扫描(full scan)和二级全扫描(product scan)总离子流图中,不得出现与土大黄苷对照品保留时间相同的离子峰。若供试品总离子流图中出现与土大黄苷对照品保留时间相同的离子峰,则核对供试品一、二级质谱图是否与土大黄苷对照品一致,如完全一致,则判定检出土大黄苷。
2.3.9 LC-MS/MS 分析
2.3.9.1 LC-MS/MS 裂解规律 按照上述色谱及质谱条件测定分析,土大黄苷对应的一级总离子流图中检出分子离子峰[M+H]+(m/z=421.2)及[M+Na]+(m/z=443.1),经对[M+H]+(m/z=421.1)准分子离子进行二级质谱子离子扫描,检出碎片离子峰[M-glu+H]+(m/z=259.1)及m/z=227.0、199.1、181.1、135.0、107.1,其中m/z=259.1和m/z=199.1 丰度比较高,选定为定性离子对。
2.3.9.2 LC-MS/MS 测定结果 一级全扫描(full scan):按上述方法测定,土大黄苷对照品一级全扫描总离子流图中在保留时间为2.5 min 时检出分子离子峰[M+H]+(m/z=421.2);供试品溶液、阳性对照溶液均检出与土大黄苷对照品保留时间相同的分子离子峰[M+H]+(m/z=421.2);阴性对照溶液未检出与土大黄苷对照品保留时间相同的离子峰。见图14至图18。

图14 土大黄苷对照品的一级TIC 图及质谱图Fig.14 The full scan of TIC and mass spectra of rhaponticin reference

图15 供试品(批号2020030101)的一级TIC 图及质谱图Fig.15 The full scan of TIC and mass spectra of sample (lot number 2020030101)
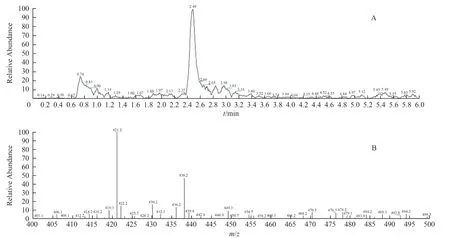
图16 阳性对照溶液的一级TIC 图及质谱图Fig.16 The full scan of TIC and mass spectra of positive control
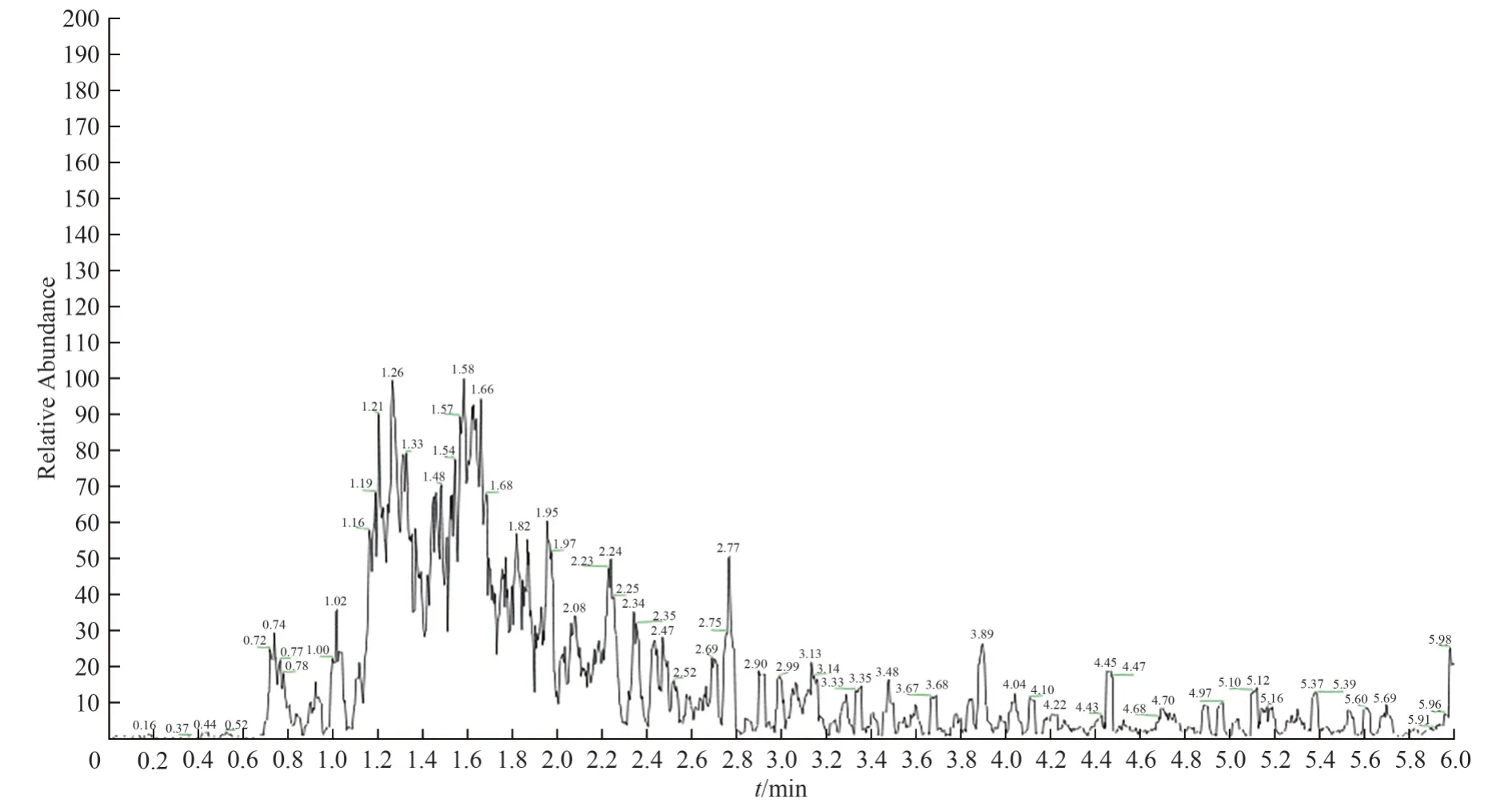
图17 阴性对照溶液的一级TIC 图Fig.17 The full scan of TIC and mass spectra of negative control

图18 空白试剂溶液的一级TIC 图Fig.18 The full scan of TIC and mass spectra of blank reagent
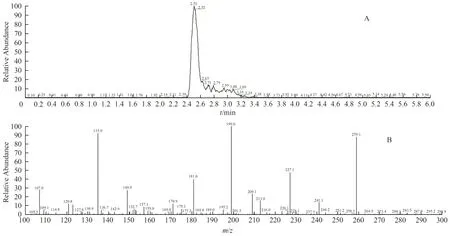
图19 土大黄苷对照品的二级TIC 图及质谱图Fig.19 The product scan of TIC and mass spectra of rhaponticin reference
二级质谱(Product Scan):对土大黄苷对照品、供试品、阳性对照溶液进行二级质谱分析,均检出保留时间为2.5 min 的分子离子峰,且二级质谱碎片质量分数和丰度比基本一致,主要二级碎片离子均包括[M-glu+H]+(m/z=259.1)及m/z=227.0、199.1、181.1、135.0、107.1。见图20 至图22。

图20 供试品(批号2020030101)的二级TIC 图及质谱图Fig.20 The product scan of TIC and mass spectra of a sample (lot number 2020030101)

图21 阳性对照溶液的二级TIC 图及质谱图Fig.21 The product scan of TIC and mass spectra of positive control
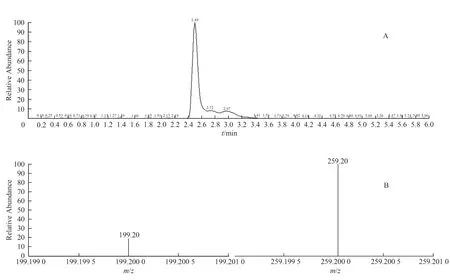
图22 土大黄苷对照品的定性离子扫描TIC 及质谱图Fig.22 The MRM of TIC and mass spectra of rhaponticin reference
定性离子对选择扫描(MRM):对土大黄苷对照品、供试品、阳性对照溶液进行定性离子选择扫描,分子离子峰m/z=421.2,二级质谱碎片离子m/z=259.1和m/z=199.1。结果均检出保留时间为2.5 min 的分子离子峰,且选择碎片离子丰度比基本一致。见图23 至图24。
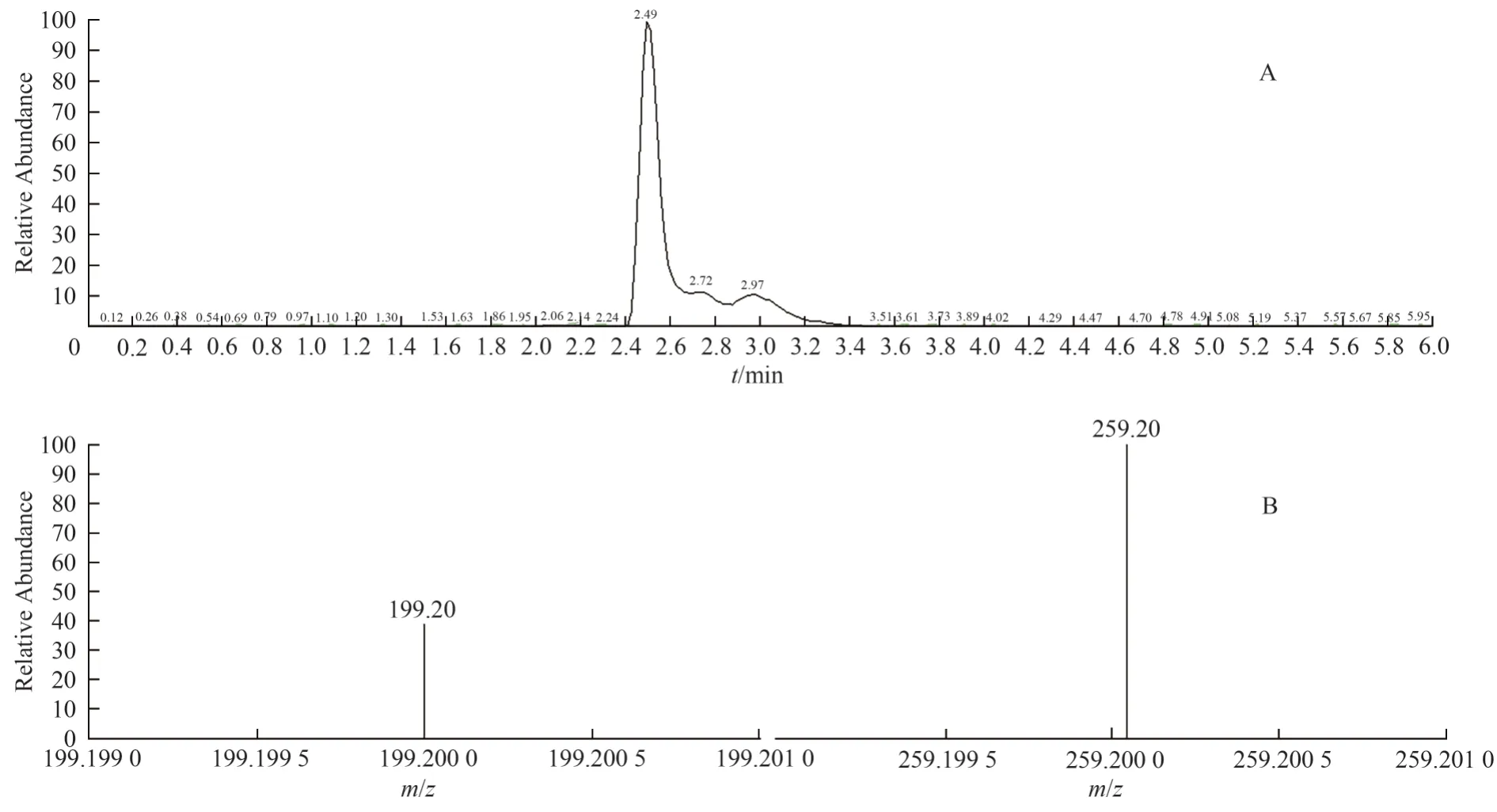
图23 供试品(批号2020030101)的定性离子扫描TIC 及质谱图Fig.23 The MRM of TIC and mass spectra of a sample (lot number 2020030101)

图24 阳性对照溶液的定性离子扫描TIC 及质谱图Fig.24 The qualitative ion scan and mass spectra of positive control
2.3.9.3 液质联用法验证的结果 根据建立的液质联用方法,12 个批次的供试品全部检出与土大黄苷对照品保留时间相同的分子离子峰,一、二级质谱图基本一致,碎片离子质量分数和丰度比基本一致;同时用定性离子对选择扫描进一步验证,也均检出相应的分子离子峰,呈阳性检出,结果与薄层色谱、液相色谱结果一致。
对此次六味能消制剂抽检涉及的各厂家提供的大黄药材用了相同的液质方法进行验证,结果各厂家的大黄药材中均未检出与土大黄苷相应的离子峰。
3 讨论
3.1 样品检测结果分析
用所建立的方法对4 个企业101 批次六味能消丸(胶囊、片)进行检测,其中A 企业生产的12 批次六味能消丸中检出土大黄苷。结果表明,部分企业在生产六味能消制剂过程中存在大黄药材未按处方规范投料的问题,表明A 企业的大黄药材质量可能存在严重问题。生产企业一方面可能由于价格等因素,为追求利益,降低生产成本而使用伪大黄投料;另一方面对药材的质量重视不够,未按《中国药典》或企业内控标准对购进的大黄进行全检,用含土大黄苷的非药用大黄代替大黄投料。生产企业在投料中使用非药用大黄投料,势必将造成质量和安全风险,影响药品疗效。
3.2 意见与建议
由于现行六味能消丸(胶囊、片)标准中无针对大黄中土大黄苷的薄层鉴别或含量测定等检验项目,造成部分生产企业对大黄药材质量不够重视,故存在以次充好的可能性。因此,建议在现行六味能消丸(胶囊、片)的标准中增加土大黄苷检查项,以保证该品种生产规范、质量可控,防止有问题的药品流入市场。本研究建立的方法已申报国家药品监督管理局,拟作为六味能消丸(胶囊、片)中土大黄苷检查项补充检验方法。
