Addressing cation mixing in layered structured cathodes for lithium-ion batteries: A critical review
2024-01-27JingxiLiGemengLingWeiZhengShilinZhngKennethDveyWeiKongPngZipingGuo
Jingxi Li,Gemeng Ling,Wei Zheng,Shilin Zhng,Kenneth Dvey,Wei Kong Png,Ziping Guo,***
a School of Chemical Engineering & Advanced Materials,The University of Adelaide,Adelaide,Australia
b Institute for Superconducting & Electronic Materials,University of Wollongong,Wollongong,NSW,Australia
Keywords: Cation mixing Layered oxide cathodes Lithium-ion batteries Electrochemical performance
ABSTRACT High-performance lithium-ion batteries (LIB) are important in powering emerging technologies.Cathodes are regarded as the bottleneck of increasing battery energy density,among which layered oxides are the most promising candidates for LIB.However,a limitation with layered oxides cathodes is the transition metal and Li site mixing,which significantly impacts battery capacity and cycling stability.Despite recent research on Li/Ni mixing,there is a lack of comprehensive understanding of the origin of cation mixing between the transition metal and Li;therefore,practical means to address it.Here,a critical review of cation mixing in layered cathodes has been provided,emphasising the understanding of cation mixing mechanisms and their impact on cathode material design.We list and compare advanced characterisation techniques to detect cation mixing in the material structure;examine methods to regulate the degree of cation mixing in layered oxides to boost battery capacity and cycling performance,and critically assess how these can be applied practically.An appraisal of future research directions,including superexchange interaction to stabilise structures and boost capacity retention has also been concluded.Findings will be of immediate benefit in the design of layered cathodes for high-performance rechargeable LIB and,therefore,of interest to researchers and manufacturers.
1.Introduction
Lithium-ion batteries(LIB)are important to energy storage because of their relatively long service life,low cost,relatively high capacity and desirable rate capability[1–3].The essential components of LIB include anode,cathode,separator and electrolyte.Currently,practically promising anodes including,graphite [4],porous carbon [5]and porous silicon [6],are attractive to researchers because of their potential for high capacity.,It is widely acknowledged however that cathode materials limit performance.Research is therefore directed to find alternate cathode materials that will exhibit greater capacity and energy density.Different types of cathodes have been proposed [7–9]including,spinel-structured LiMn2O4(LMO)[10],olivine-type LiFePO4(LFP) [11]and layered structure oxides(LiTMO2,TM=transition metals)[12].
The cathodes are negatively affected by the antisites defects caused by cation mixing and correspond to site exchange between TM and Li ions.For example,Hsu et al.reported 2.81% Li+exchanged sites with Fe2+in LFP [13].In olivine-type cathodes,site change behaviour has been reported to restrict Li+diffusion pathways,that critically limits battery capacity [14].Zou et al.reported 4.2% Co/Li cation mixing in LiCoPO4cathodes,and 2.3% antisite defects in carbon-coated samples where the bare LiCoPO4exhibited just 121.3 mAh g-1initial discharge capacity(134.7 mAh g-1for the modified sample) [15].Additionally,using the X-ray diffraction technique,Tarascon et al.reported Li/Mn mixing can reach 10% in LiMn2O4powders[16].
The history of layered oxides in battery research dates to the 1970s when TiS2and TaS2were proposed as cathodes with Li insertion/extraction during battery discharging/charging [1].Later,electrochemical reactions for LiCoO2(LCO) at high temperature were determined.Subsequently,Sony® commercialised LCO with graphite counter-electrodes in 1991[17].However,an accurate characterisation for Li/Co cation mixing was incomplete.Wang et al.reported Co ion migration in LCO.Migration results in the formation of rock salt and spinel phase in the cycled LCO that limits capacity retention to just 86.7%after 50 cycles [18].Lithium/manganese cation mixing in manganese-based cathodes is similarly important.For example,LiMn2O4-type spinel phase was reported in LiMnO2layered oxides,that resulted from phase transition because of Li/Mn mixing [19].The most extensively reported Li/Ni mixing is a common phenomenon in Ni-based layer cathodes.Yoon et al.reported 2% Li/Ni antisite defects in LiNiO2cathode.A rock salt phase at the surface was reported in cycled electrodes and Li/Ni cation mixing in LiNiO2confirmed to intensify during battery operation [20].Research interest has also focused on ternary cathodes with high energy density,reasonable cycle stability and reduced toxicity.Therefore,LiNi1/3Co1/3Mn1/3O2(NCM333),LiNi0.5-Co0.2Mn0.3O2(NCM523) [21],LiNi0.5Mn0.5O2(NM55),LiNi0.6-Co0.2Mn0.2O2(NCM622) [22],LiNi0.8Co0.1Mn0.1O2(NCM811) [23],LiNixCoyAl1-x-yO2(NCA) [24]and other,Ni-rich ternary cathodes LiNix-CoyMnzO2(0.8
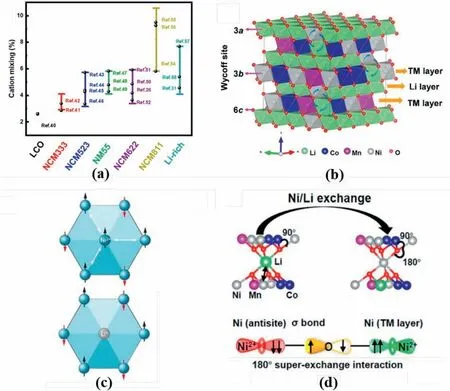
Fig.1.(a) Cation mixing degree in different cathodes.Data were extracted from Refs.[26,31,40–58].(b) Schematic for crystal structure for R-3m NCM.Adapted from Ref.[39].Copyright 2016 The Authors.(c) Schematic for TM at 3b sites in TM layer of R-3m structure,together with spin direction for each atom.Reproduced with permission from Ref.[44].Copyright 2020,American Association for the Advancement of Science. (d) Schematic for formation of superexchange interaction in R-3m structure.Reproduced with permission from Ref.[41].Copyright 2017 American Chemical Society.
In 2019,Pan et al.reviewed Li/Ni disordering in layered oxides with a focus on formation mechanisms,electrochemical impact,and corresponding modification controls[33].More recently,Zheng et al.reported on Li/Ni mixing,especially in Ni-rich layered materials [34].Importantly,however,the critical Li/Mn and Li/Co antisites in the layered cathodes were not reported on by these authors,resulting in a limited treatment of critical underlying mechanisms for cation mixing in layered oxides.A comprehensive review that addresses Li/Ni,Li/Co and Li/Mn mixing is needed to aid the development of high-performance layered cathodes for next-generation LIB.Here,we review the cation mixing behaviours in the layered cathodes with the start of their formation mechanisms in the structure.Meanwhile,corresponding advanced techniques to characterise cation mixing are systematically summarised,highlighting the working principles as well as their strengths and shortcomings.The critical roles of cation mixing on the battery performance of the layered oxides has been revealed from both structural and kinetic aspects,which sheds light on the underestimated benefits of cation mixing and refreshes the previous biased stereotype in most relevant publications.Subsequently,up-to-date advances on regulating the degree of cation mixing in layered oxides are included,covering the calcination parameter optimisation,elements substitution and surface engineering.Lastly,we provide our perspectives on the future direction of cation mixing related research in layered cathode materials for LIB.This work will contribute to the revaluation of the roles of cation mixing and accelerate the development of stable and high-capacity layered oxides cathodes.
2.Origin of cation mixing in layered cathode materials
It is widely accepted that cation mixing is closely related to electrochemical performance [35,36].In the α-NaFeO2type layered NCM material withR-3mspace group symmetry in Fig.1b,Li,TM,and O reside at 3a,3b,and 6cWyckoff sites,respectively,forming the alternation between the Li and TM layers in the rhombohedral[001]direction[37–39].However,this structure is interrupted by cation exchange known as Li/(Ni,Mn,Co)antistites(Fig.1b).Li+diffusion into the TM layer with synchronous TM migration into the Li layer occurs during heat treatment and electrochemical reaction.
Significant research has been directed to confirm the origins of the Li/Ni mixing in the Ni-rich layered cathode [40,41],including 1) Li+and Ni2+have similar bonding environments in theR-3mstructure,namely,both ions reside at the interstitial sites of the oxygen octahedra and edge-sharing with the adjacent octahedral [42],2) the similar ionic radius of Li+(0.76 Å)and Ni2+(0.69 Å)contributes to the formation of the Li/Ni antisites defects [43],3) the Li/Ni antisites contributes to the entropy reduction of the system and structure stabilisation,4) Li/Ni mixing reduces ‘magnetic frustration’ of the structure [44],and 5) the phase evolution induces Li/Ni mixing during the electrochemical reactions[45].
It is confirmed that the formation mechanism of a honeycomb-type superstructure is observed in the TM layers for the layeredR-3mstructure (Fig.1c).Given the relatively strong magnetic moment of Ni,the edge located Ni2+in the honeycomb superstructure should have different magnetic moments from neighbouring ones,resulting in the triangular configuration and concurrent creation of magnetic frustration in the structure.This worsens when a centred Ni2+is added into the superstructure,leading to unstable high-energy states of the layered cathodes.Because Li ions have no magnetic moment,the spin loss at one site caused by the Li/Ni exchange significantly reduces structural magnetic frustration,leading to Li/Ni antisites defect formation.
Pan et al.[41]reported the critical role of the superexchange interaction in tuning the Li/Ni antisites defects in the layered cathodes(Fig.1d).Based on theoretical calculations,the Li/Ni mixing in NCM materials results in linear antiferromagnetic Ni2+-O2--Ni2+superexchanges because of the spin-flip for Ni2+in the Li layer.Greater Ni concentration,especially Ni2+,in NCM cathodes leads to increased Li/Ni antisites mixing in the layered structures[42].These authors claim that Ni3+in NCM materials has priority to exchange with Li+,which is subsequently followed by a reduction in valance state and formation of antiferromagnetic Ni2+-O2--Ni2+superexchanges.The Ni3+/Ni2+reduction contributes to oxidation in the neighbouring Co,i.e.,Co4+/Co3+,because of the charge compensation.The Co4+forms new,linear interlayer ferromagnetic Ni2+-O2--Co4+and intralayer antiferromagnetic Co4+-O2--Ni2+/Ni3+superexchanges,increasing Li/Ni mixing in the structure.
Li/Ni mixing during prolonged electrochemical cycling is reported by Wang et al.in which the mixing degree of bare-NCM622 increased from 4.87 to 9.02% after 100 cycles[46].The reason hypothesised is that the low migration energy barrier of the Ni ions.Two Ni ion diffusion pathways have been hypothesised: 1) as illustrated in Fig.2a,the Ni2+can move from its octahedral siteviathe tetrahedral site to the octahedral site of Li+vacancy,named Oh→Td→Oh[47]and 2)alternatively,through the O vacancies,the Ni ion in the transition layer octahedral site migrates to the Li layer octahedral site named Oh→Vo→Oh(Fig.2b)[47].The energy barrier for the two routes has been calculated by Kim et al.,as is shown in Fig.2c and d.The energy barrier for Oh→Td→Ohis 0.560–0.598 eV,and for Oh→VO→Oh,0.567–0.617 eV[47].The lower energy barrier for Ni to diffuse into the Li layer causes performance degradation.

Fig.2.Schematic for Ni ion migration pathways with (a) Oh→Td→Oh and (b) Oh→VO→Oh.Calculated energy barriers for (c) Oh→Td→Oh and (d) Oh→VO→Oh at delithiation stages,where the x-axis (abscissa) corresponds to positions in (a) and (b).Reproduced with permission from Ref.[47].Copyright 2011 American Chemical Society. (e) Atomic resolution Scanning Transmission Electron Microscope -Electron Energy Loss Spectroscopy mapping for TM ions from layer to bulk.Reproduced with permission from Ref.[48].Copyright 2017 American Chemical Society.(f)Calculated formation energy for possible migration mechanisms for TM ions in bulk.Reproduced with permission from Ref.[49].Copyright 2017 American Chemical Society.
Other TM/Li disordering has been observed in tannery cathode materials,and the exchange principle is confirmed.In contrast to Ni ions that exchange with Li during electrochemical cycles and calcination,other TM ions tend to exchange with Li in the Li layer because of the lower energy barrier induced by the reduction of the valance state[50].The high-angle annular dark-field scanning transmission electron microscopy(STEM-HADDF)imaging was used to detect cation mixing in the NCM333(Fig.2e)[48],where TM ions migrate from the TM layer to the Li sites in the Li layer.The disordered cation distribution of Ni was observed together with the migration of Mn and Co in the HADDF images(Fig.2e).At the highly delithiated state,Co and Mn ions migrate to the vacancies in the Li layer,which results in the occupation of Li+at the octahedral sites of the TM layer during lithiation,and the formation of Li/Co and Li/Mn cation mixing in the structure(Fig.2e)[49].The Li/Mn and Li/Co cation mixing is intensified in the extensively cycled electrodes.Therefore,a detailed assessment of Li/Mn and Li/Co cation mixing is needed.For example,the formation energy for the antisites defect of Li/Mn,Li/Co and Li/Ni has been evaluated via simulation for different electrochemical procedures.As is displayed in Fig.2f,at different delithiated states the Li/Ni antisite formation energy is lowest,whilst the Li/Mn antisite is the most difficult to form[49].
3.Characterisation method of cation mixing
Given the close relation of cation mixing with performance,it is necessary to characterise Li/TM ions antisites and quantitatively determine the degree of mixing of the layered structure.Several techniques are used,including X-ray/neutron powder diffraction and electron microscopy.Here,different characterisation tools and discuss the working principle will be compared in detail to emphasise the importance of precisely characterising the cation mixing.
3.1.Powder diffraction
Powder diffraction was devised in the 1910s and continued in use in material research into the 1970s.With the introduction of the Rietveld refinement method,researchers determine 3D-dimensional structural details of materials from one-dimensional powder diffraction data[51].Because powder diffraction provides mean structural information of the powder data for cation mixing in the layered materials can be reliably interpreted at a global level,i.e.,average information.
3.1.1.X-ray powder diffraction (XRPD)
Laboratory XRPD is the most widely used diffraction to detect cation exchange in the layered structure [52,53]because of its high accessibility,(moderately) facile operation,and low characterisation requirements.
In principle,X-rays only interact with the electrons of the elements[54],resulting in an atomic-number-dependent scattering factor and a high contrast between Li and TM ions.For example,a direct indicator for the occurrence of Li/Ni mixing from the XRPD data is the integrated intensity ratio between 003 and 104 reflections,which have been widely used[52,53,55,56].The schematic for the structure of NCM is shown in Fig.3a,with its(0 0 3)and(1 0 4)lattice planes indicated.Obviously,theR-3m(0 0 3) plane is filled with transition metal ions,while theR-3m(104)plane involves both Li and transition metal.The exchange of Ni at 3bsites and Li at 3asites in the structure significantly change the intensity ratio evolution of peak003/peak104.Therefore,the intensity ratio can be used to reflect the degree of Li/Ni mixing.Using NCM333 as an example,the simulated XRPD patterns of theR-3mNCM with different Li/Ni mixing degrees are shown in Fig.3b.The integrated peak intensity of 003 reflection decreases monotonously with the increase of Li/Ni exchange,while no significant changes are observed for 104 reflections(Fig.3c).Generally,it is widely accepted that an intensity ratio value<1.2 from the XRPD pattern indicates cation mixing in the layered materials[2,52],leading to unsatisfactory battery performance.In addition to(0 0 3)and(1 0 4)reflections,the(1 0 8)and(1 1 0)reflections are also employed to institutionally determine the Li/Ni mixing degree,where the merge of these two peaks is usually associated with the high-concentration Li/Ni antisite defects[52].With the development of synchrotron-based instrumentation,synchrotron-based XRPD has also been widely employed as an advanced XRPD tool to probe the internal Li/Ni exchange of layered NCM cathodes[57,58].In addition to the high resolution and high brilliance,the tuneable wavelength of the synchrotron-X-ray beam further contributes to its popularity in structural characterisation.
3.1.2.Neutron powder diffraction (NPD)
Unlike that XRPD interacts with electrons,NPD involves the interactions between atomic nuclei,thus endowing NPD with unique insights into the structural information of battery materials,complementary to that gained from XRPD[54,59].The neutron coherent scattering length (b) is independent of the element atomic numbers,which contributes to its high sensitivity to light elements,especially Li and O,and distinguishes between elements with similar atomic numbers[54,60].For instance,in the NCM cathodes,the elements(Li,Ni,Mn,Co and O) havebvalues of -1.90,10.3,-3.73,2.49,and 5.80 fm(1 fm=10-15m),respectively,showing comparable contrast in the NPD data.
NPD patterns of NCM333 with different Li/Ni mixing degrees were simulated,with the profile shown in Fig.3d.The peak intensity decrease with the rise of Li/Ni exchange has been observed for the reflections of(003),(101),(107),etc.The Li/Ni mixing degree in the layered NCM materials could be precisely and quantitively determined using NPD data using the Rietveld refinement method.
As is demonstrated,publications use the peak intensity ratio of(003)/(104) to represent Li/Ni mixing degree qualitatively.When the value is>1.2,the mixing level is described as ‘acceptable’.However,the intensity ratio cannot accurately represent cation mixing degree,and distinguish separately Li/Ni,Li/Co and Li/Mn cation mixing.Additionally,peak intensity is significantly affected by equipment,and test conditions.Therefore,a quantitative determination of the cation mixing degree is required.The Rietveld refinement,or Pawley analysis on powder diffraction data quantitatively characterizes cation-mixing degree in different cathodes.Because of different coherent scattering lengths of TM ions,refinement results on NPD data accurately confirm degree of Li/Mn,and Li/Co cation mixing.Therefore,it is recommended to use refinement analyses for powder diffraction data to characterise cation mixing quantitatively.
3.2.Electron microscopy technique
Both annular bright-field (ABF) and high-angle annular dark-field(HAADF) modes of the aberration-corrected scanning transmission electron microscopy(STEM)technique could be employed to investigate the atomic structures of the layered cathodes.Typically,HAADF STEM mode employs the annular detector with an inner angle much larger than the probe-forming aperture convergence angle.ABF STEM mode uses an outer angle commensurate with the probe-forming aperture convergence angle [61].In the HAADF STEM mode,the signal intensity is proportional to the atomic numbers(Z)of elements,where only heavy elements(Ni,Mn,Co,etc.)are visible in the appearance of bright columns[62,63].By contrast,the ABF STEM mode shows an inversely proportional Z-dependent intensity [61].However,it is still challenging to directly visualise lithium in the structure using STEM [56].In most circumstances,ABF STEM images are complementary to the HAADF STEM mode.Since only tiny areas can be detected,the information obtained from STEM would be at a localised level (short range).
For example,Yang et al.[63]showed the Li/Ni antisite defects formation in the NCM333 cathodes during charging.The schematic structure of pristine NCM333 on the[010]projection,which enables the Li,O,and TM columns to be well aligned,is shown in Fig.4a,with the corresponding HAADF and ABF STEM images shown in Fig.4b and c,respectively.In Fig.4d,the TM atoms at 3bsites are observed as bright dots.In Fig.4c,the Li and O atoms at 3aand 6csites are shown as dark holes.Line profile analysis along the [42–1]direction of theR-3mstructure was carried out to obtain further information on the atomic configuration within the structures,where it is noted that the peak intensity correlates with Z of elements.The profile in Fig.4d and e demonstrate a periodical distribution of the TM-O-Li-O-TM ordering,corresponding to a perfect layeredR-3mstructure without noticeable Li/Ni exchange.Fig.4f and g shows the corresponding HAADF and ABF STEM images of NCM333 cathodes at full charge,in which the layered structure is well maintained.With careful investigations,one can find that some atoms reside between the adjacent TM slabs at the surface of the particles(Fig.4f),which corresponds to the antisite occupation of Ni and Li at 3aand 3b,respectively.Meanwhile,the occurrence of Li/Ni exchange in the structure is further confirmed by the ABF STEM results in Fig.4g.
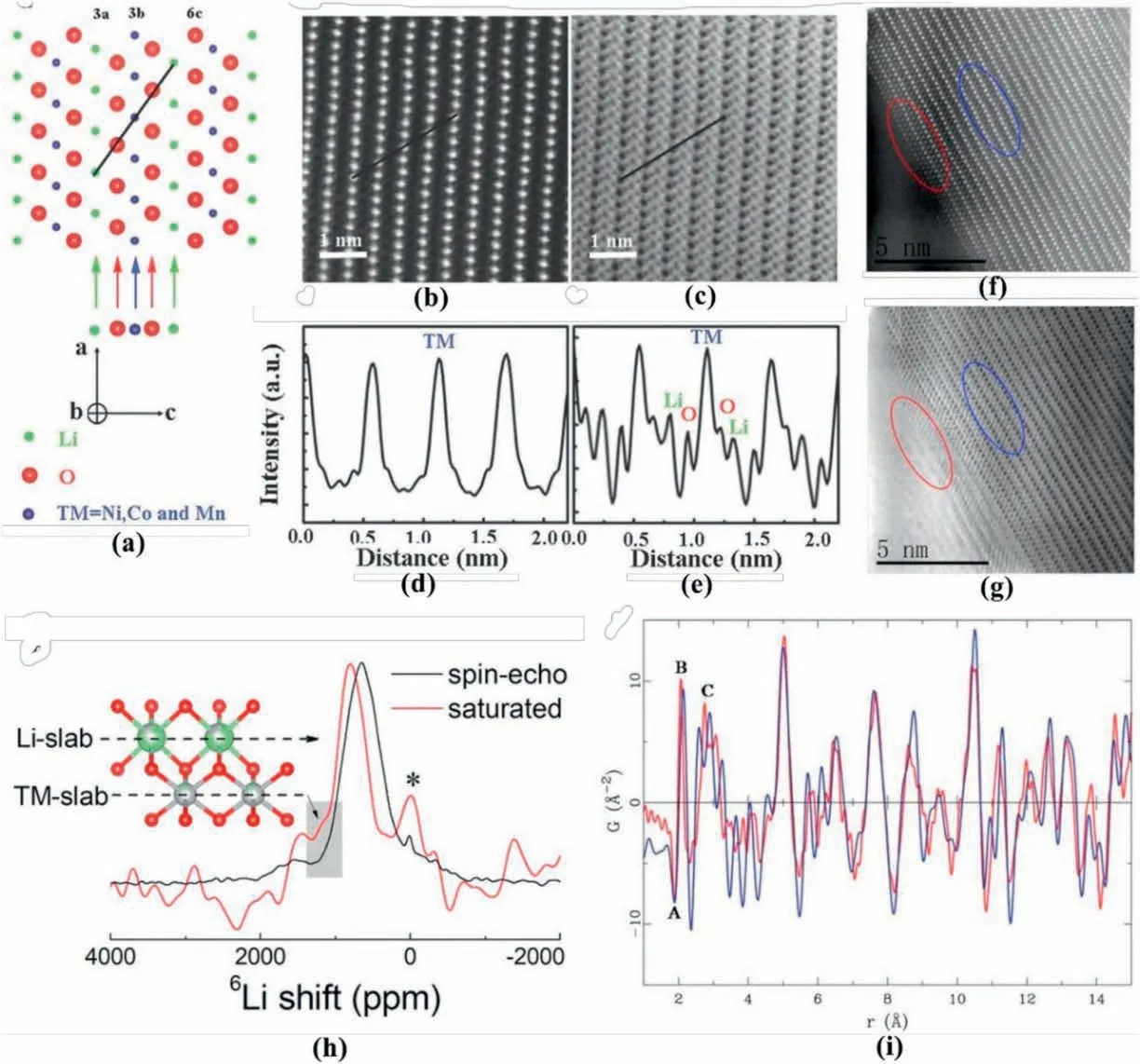
Fig.4.(a) Schematic for R-3m NCM, (b) HAADF and (c) ABF STEM images of pristine NCM from [010]direction,together with (d) and (e) corresponding profile analyses,(f)HAADF and(g)ABF STEM images of NCM material at full-charge.Reproduced with permission from Ref.[63].Copyright 2016 Wiley-VCH Verlag GmbH&Co.KGaA,Weinheim.(h) 6Li NMR spectra for layered LiNi0.7Co0.3O2 materials at the spin-echo sequence and saturated states.A schematic was added to show the signal sources.Reproduced with permission from Ref.[64].Copyright 2019 American Chemical Society. (i) PDF profile for layered LiNi0.5Mn0.5O2 prepared by solid-state route (red-colour curve) and ion-exchange method (blue curve).Reproduced with permission from Ref.[65].Copyright 2007 The Royal Society of Chemistry.
3.3.Other potential tools
In addition to the abovementioned techniques,other advanced technologies,which include solid-state nuclear magnetic resonance(NMR) and pair distribution function (PDF) [64–67],have also been adopted due to their sensitivity to cation exchange.
Solid-state NMR has a high sensitivity to lithium's local environment and the corresponding chemical shift resulting from the influence of surrounding paramagnetic TM ions [66].For example,Wang et al.[64]employed6Li NMR to detect the Li/Ni antisite defects in the layered LiNi0.7Co0.3O2cathode,with the data profile shown in Fig.4h.Considering the relaxation time difference of Li at different sites(i.e.3aand 3bsites,respectively),spin-echo sequence and reversion recovery sequence with a recovery time of 0.012s were applied during NMR experiments to saturate Li at the 3asites and suppress its signal,thus resulting in the highlighted signal reflecting 3b-located Li.
Typically,in the6Li NMR spectra,only two types of peaks are spotted,including one coming from the overlapped Li signal in theR-3mstructure and the other(at~400 or 300–1200 mg/L,respectively)resulting from the undesirable Li2CO3/LiOH impurities [64].After saturation,a weak shoulder peak can be found at the low field side (around 1260 ppm,as shown in the shaded box in Fig.4h in the LiNi0.7Co0.3O2NMR spectra,revealing the occurrence of Li exchanging with Ni in the structure.Meanwhile,this characteristic small and broad peak indicates the tiny amount of the Li/Ni anti sites in the structure.
Similar to NMR,the PDF technique is also powerful in detecting the local structure of battery materials.Unlike traditional X-ray/neutron diffraction methods involving only Bragg scattering,PDF analysis considers both Bragg scattering and diffuse scattering[68].The PDF G(r)can be obtained from the diffraction data after performing a Fourier transform on the normalised scattering intensity[65,68].Generally,PDF G(r)is regarded as a distribution function of various bond lengths,reflecting the probability of finding an atom at a certain distance from another atom.For example,Grey et al.[65]compared the neutron-based PDF data of different LiNi0.5Mn0.5O2prepared by ion-exchange and solid-state methods to investigate the structural difference.The corresponding data for LiNi0.5Mn0.5O2are displayed in Fig.4i.Peak A appears as the first correlation distance,which corresponds to the bonds between Mn4+and O and owns a negative peak intensity due to the negative scattering length of Mn (-3.73 fm).Peak B is located at around 2.09 Å and is attributed to Ni–O and Li–O bonds,due to the close bond length of Ni–O and Li–O pairs and difficulty distinguishing them.Peak C stands for the close contact of TM-TM,TM-O,and O–O pairs.The difference in intensity of peak B evidences different Li/TM distributions in the structure.Via the Rietveld refinement of neutron data,reverse Monte Carlo computations confirmed the Ni coordination environment,and therefore,different Li/Ni distributions in the structure.Because of the characterisation on TM-O bond length,PDF findings combine with STEM imaging.Via comparing the bond length in the STEM image and PDF findings,the cation mixing behaviour is illustrated.
In summary,various cutting-edge techniques could be employed to detect cation exchange in the layered structure.Although XRPD is popular and powerful,its insensitivity to Li and weak ability to distinguish transition metals should be considered when exploring the exact level of Li/TM in the structure.As for NPD,its accessibility would probably limit its wide application.It is noted that the combination of XRPD and NPD analysis would be an excellent practice to probe cation mixing and other structural details.For the local-focused techniques,especially electron microscopy,a multi-site investigation should be conducted to obtain feasible and convincing conclusions.For NMR and PDF techniques,more developments need to be made in the future to not only qualitatively but quantitively detect the Li/TM exchange in the layered NCM cathodes.Finally,considering the possibility of a discrepancy between conclusions from electrode-level (~50 μm) and local-level (1–5 nm) observations,more characterisation techniques are required,especially those reflecting the desired structural information at the transitional particle-level (~1 μm).
4.Consequence of the cation mixing
4.1.Influence on phase evolution and electrochemical performance
The hazardous phase evolution induced by the antisite Ni migration in the LiNi0.5Mn0.5O2layered oxides has been reported[69].Calculation results showed that the migration energy barrier of the Ni ion in the Li layer is merely 0.25 eV,significantly lower than the Ni migration energy barrier in the TM layer (1.47 eV).The results indicated that the Ni ions were easier to migrate in the Li layer than in the TM layer.Therefore,the spinel or rock-salt phase could be formed more easily when the amount of antisite Ni is higher[69].For example,the existence of a rock-salt phase on the severely cycled LiNi0.7Co0.15Mn0.15O2(NCM71515)has also been confirmed[70].A deeper rock-salt phase(200 nm)was identified at the surface region of the 1500 cycled samples.Moreover,the migration of the TM ions and the site exchange between TM/Li induce the formation of undesired phase especially at the surface region of the particles.It is reported that Li/Ni mixing in NCM811 could result in the transition from the layered structure to the rock salt phase [71].The cycled NCM811 features a rock-salt structure at the particle surface caused by the Li/Ni mixing,as shown by HADDF STEM images in Fig.5,while its bulk structure remains layered.The surface rock-salt phase could gradually diffuse into the material bulk and finally result in the battery failure of the NCM811 materials[71].

Fig.5.STEM-HAADF images for different regions from bulk to surface.The white colour dashed lines denote surface reconstruction layer.Corresponding structural models are provided below.Reproduced with permission from Ref.[71].Copyright 2018 Elsevier Ltd.
In LiMnO2layered cathodes,the structure degradation resulting from Li/Mn cation mixing significantly affects electrochemical performance.During the lithiation/delithiation process,Mn ions migrate to the Li layer vacancies and occupy the octahedral sites [19].When Li+inserts back into the layered structure,the Mn migration-induced vacancies in the TM layer are occupied by Li+,leading to the phase transformation from layered to spinel LiMn2O4-type[19].The Li/Mn cation mixing results in irreversible phase evolution and the loss of Li+active sites that deteriorates battery capacity and cycle stability of active materials.Moreover,Li/Mn antisite defect has been reported in Li1.2Mn0.55Ni0.15Co0.1O2oxides to form the LiMn2O4-type spinel phase,which evidenced that Li/Mn cation mixing affects the layered structure significantly[72].
In the layered cathodes,the Co ion migration induces the formation of undesired rock salt and spinel phase.Co ions can be stabilised with a tetrahedral coordination environment,given the electronic configuration of Co2+as[Ar]3d7.In the LCO structure,Co ion migration leads to both octahedral and tetrahedral occupation of Co within the layered structure,resulting in the formations of rock salt and Co3O4-type spinel phases that limits capacity retention to just 86.7% after 50 cycles [16].Migration of Co ions in the Ni-rich LiNi0.83Co0.11Mn0.06O2was reported[68].As Co occupies tetrahedral sites that serve as an intermediate station for the migration of Ni and Mn,the Li/Ni and Li/Mn cation mixing in the structure is relieved[68].
However,the influence of cation mixing on phase evolution is still debating.Cation mixing could tune the mechanistic phase evolution of the electrode materials upon electrochemical reactions.For example,Nirich cathode materials usually suffer phase transformations between H2 and H3 (hexagonal phases with different lattice parameters) during charge/discharge (Fig.6a) [73].When delithiated at 4.2 V,the active material experiences a dramatic shrinkage along thecaxis of the layered structure,which result in abrupt lattice contraction,anisotropic strain within material grains,severe particle pulverisation and eventually failure of the active material[74].Wu et al.introduced Ti dopants into the Li sites of LiNi0.9Co0.1O2,which increased the Li/TM cation mixing at the surface layer of the material particles.The antisite TM ions in this surface layer could restrict the H2/H3 phase transformation in the surface region,which diminishes the propagation of the undesirable phase transformation from surface to bulk,as evidenced by the observations ofin-situXRPD,HRTEM,and SEM characterisation in Fig.6b [75].Accordingly,the capacity retention was improved from 69.68 to 97.96% under 0.2 C after 100 cycles,and the rate capability was enhanced to 143.4 mAh g-1at 10 C.
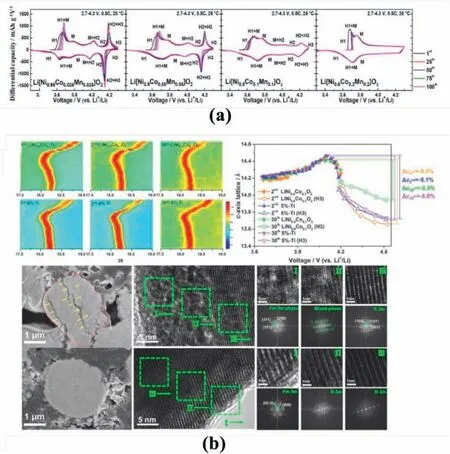
Fig.6.(a) Phase transformation from dQ/dV curves for Ni-rich cathode material with different Ni content.Reproduced with permission from Ref.[73].Copyright 2018,American Chemical Society. (b) In-situ XRD patterns for,and HRTEM images of,LiNi0.9Co0.1O2 and Ti-modified samples illustrating suppression of H2 to H3 phase transformation.Reproduced with permission from Ref.[75].Copyright 2019 Published by Elsevier Ltd.
4.2.Restriction on Li ion diffusion
During sintering,cation mixing occurs inevitably due to the similar ionic radius of Ni and Li and the relatively low energy barrier for Ni2+to migrate [64,76].The antisite cation could hamper the transportation of the Li ions in the layered cathodes during charge/discharge,thus deteriorating the charge/discharge capacity and rate capability of the active material.As shown in Fig.7,the Li+diffusion can be categorised into two models: oxygen dumbbell hopping (ODH) and tetrahedral site hopping(TSH).In the model of ODH,Li+migrates from one octahedral site to a neighbouring octahedral site through the oxygen dumbbell.Whist in TSH,Li+diffuses by way of the adjacent tetrahedral site.It is found that ODH is preferable at the beginning of the charging process(delithiation),and TSH dominates when more than 1/3 Li ions are extracted from the structure[77].

Fig.7.Schematic for kinetics of Li-diffusion in delithiation.Background colours (orange and green) illustrate dominant area for,respectively,TSH and ODH.Reproduced with permission from Ref.[77].Copyright 2015,American Chemical Society.
Compared with the Li+,antisite TM ions have a higher valance state,which showed a stronger electrostatic repulsion to the moving Li+,which suppress Li mobility when the Li+transfer alongside the ODH pathway[78].Secondly,TM ions occupying Li sites act as an obstacle in the 2D Li diffusion pathway to block Li migration.As the active Li diffusion sites were occupied,ODH and TSH diffusion has been restricted [77].Moreover,as the TM-O bond energy is higher than the Li–O,the Li slab distance will be narrowed down,no matter whether the ODH or TSH diffusion could be influenced due to the increase of the activation barrier of the Li diffusivity.At last,after the first charge happens,the Ni2+will be oxidised to Ni3+,a local shrinkage will rise around the Ni ion,and the irreversibility of the first cycle will be accounted for,which is shown as the lithium ions are difficult to insert back[79].All above mentioned will account for the reduction in Li+diffusion efficiency.Consequently,the impact on Li diffusion is reflected in the electrochemical performance of the materials.For instance,as reported by Gao et al.,the modified NCM333 with lower Li/Ni mixing degree demonstrated an improved Li diffusion coefficient of 3.765×10-11cm2/s(vs.4.621×10-12cm2s-1),a higher initial discharge capacity (197.9vs.171.7 mAh g-1),and a greater rate capability(120.9vs.49.5 mAh g-1at 8 Cvs.49.5 mAh g-1)[80].
4.3.Structure stabilisation
Despite the influence of cation mixing on phase evolution and Li+diffusivity,the contribution of cation mixing on structural stabilisation of the layered cathodes has also been confirmed.For instance,Ni ions were doped to the Li layer of LCO,artificially increasing the cation mixing degree and inducing the pillar effect of Ni at Li sites to stabilise the structure,especially at a high operating voltage.The capacity retention of LiCoO2was improved by 33.7% after 100 cycles at 1 C within a 3.0–4.5 V voltage window [81].Additionally,Zheng et al.confirmed the roles of cation mixing in layered oxides in enhancing structural stability during their investigation of the underlying superexchange mechanism [82].After the exchange of Ni ions to the antisites,a 180-degree superexchange interaction was found to form between the antisite Ni and the Ni at TM layer,which is significantly stronger than previous interactions between Li and Ni and thus boosts the structural stability.
Similarly,the antisite Co and Mn contribute to the intense interactions with TM ions in the TM layerviathe superexchange mechanism[82].Therefore,considering the strong superexchange interaction in the structure,cation mixing improves the structural stability and cycling performance of the active materials.Moreover,Delmas et al.studied the LiNiO2material through extended X-ray absorption fine structure(EXAFS),with interesting findings showing that the Ni2+in the Li slab of the structure could maintain the structural integrity by preventing the macroscopic distortion of the material particles[83].
In summary,cation mixing is closely related to the electrochemical performance of the layered cathode materials.It could be a parasitic mixing formed during the calcination process and aggravated during the electrochemical reaction due to the ion migration in the structure [84].Up to date,the impacts of cation mixing on electrode performance are controversial [40].On the one hand,cation mixing lead to low battery capacity due to the loss of Li active sites,as well as the undesirable phase evolution induced by the antisite cations.While,on the other hand,the antisite cations act as structural pillars,thus contributing to improved structural stability during electrochemical procedures.
5.The regulation of cation mixing
The comprehensive influences of cation mixing on the battery performance of layered cathode materials has been shown in the previous section.It is necessary to develop strategies to regulate the cation mixing degree during preparation and the electrochemical process.In this section,we categorise the modification strategies into sintering parameter optimisation,elemental substitution,and surface engineering with up-todate examples.Additionally,the underlying working mechanisms are also discussed in detail.
5.1.Sintering parameter optimisation
Although cation mixing occurs inevitably in the layered cathode materials,sintering parameters,such as atmosphere [85,86],temperature [87],and cooling condition [88],can be modified to regulate the degree of cation mixing in the layered oxides to achieve superior electrochemical performance.
The sintering atmosphere plays a vital role in controlling the Li/TM mixing in the layered cathodes.For example,the Li/Ni mixing degree of NCM523 sintered in different atmosphere(N2,air and 40% O2)has been compared,where the NCM523 prepared in N2showed the highest Li/Ni mixing degree of 8.3%,and the sample prepared in O2delivered only 0.6% mixing degree.The sample with the lowest Li/Ni mixing degree delivered a high discharge capacity of 176.4 mAh g-1under 2 C [85].Similarly,Yuan et al.also reported that NCM622 synthesised in O2shows a lower Li/Ni mixing (1.58%) and better electrochemical performance(e.g.184.6 mAh g-1at 0.1 C and 83.7% capacity retention after 100 cycles under 0.5 C) compared with that prepared in the air [86].
Generally,in layered cathode materials,Ni2+is prone to exchange with Li+due to the similar ionic radius,while O2atmosphere can sufficiently oxidise Ni2+to Ni3+,thereby reducing the cation mixing in the active material.Meanwhile,the O2atmosphere also reduce the formation of oxygen vacancies at the particle surface,resulting in the stabilised layer structure.Therefore,oxygen atmosphere is usually employed during the preparation of Ni-rich layered cathode materials to avoid excess cation mixing in the structure.
Calcination temperature would also impact the cation mixing degree and the structure orderness.However,due to the varieties of the precursor and the sample compositions,as well as their synthesis methods,it is too difficult to claim a proper sintering temperature and holding time.For instance,He et al.argued that the NCM811 samples sintered at 800°C for 10 h delivered the best performance alongside the lowest Li/Ni mixing(1.08%)in the structure[87].Whilst,the NCM71515 sintered at 850°C showed the lowest Li/Ni mixing and the highest discharge capability of 197 mAh g-1(Fig.8a and b)[76].

Fig.8.(a) Charge/discharge curves and (b) Cycle performance for NCM71515 sintered at different temperature. (c) Evolution of cation disordering reflected by occupation variation of Ni ions at 3b sites under different calcination temperature.Reproduced with permission from Ref.[76].Copyright 2017 Wiley-VCH Verlag GmbH&Co.KGaA,Weinheim.(d)Evolution of percentage of Ni at 3b sites as a function of temperature.Reproduced with permission from Ref.[89].Copyright 2019 Wiley-VCH Verlag GmbH&Co.KGaA,Weinheim.
Although the sintering temperature varies,prolonging the sintering time sufficiently regulate the Li/Ni mixing into a desirable degree.During the temperature holding process,the extended sintering time mitigated the migration of Ni2+from the TM site(3b)to Li site(3a)sites,thus reducing the degree of Li/Ni mixing(Fig.8c).Meanwhile,it has also been noted that high temperature and long sintering time would lead to severe lithium loss in the active material,making more active antisites for TM ions to occupy,which may cause the increase of the cation mixing degree and the loss of battery capacity [76].Moreover,cooling condition also influence the cation mixing degree.It has been reported that the quenching process would result in a higher degree of Li/Ni mixing and lower battery capacity of the active material than that processed with natural cooling (Fig.8d) [89].During the cooling procedure,Ni2+also migrates back to the octahedral site in the TM layer,which reduce the cation mixing[76].
In summary,to balance the Li/O loss and obtain a low cation mixing degree in the material structure,the sintering temperature should be reasonably low with enough ordering activation energy provided alongside an acceptably elongated holding time.Proper cooling control and O2atmosphere are also vital to obtain the layered cathodes with desired cation mixing.It is vital to take all these factors into careful consideration,prior to the fabrication of the layered cathode materials to achieve a superior overall electrochemical performance.
5.2.Elemental substitution
Introducing the foreign elements to the host structure has been extensively employed to enhance the overall electrochemical performance of the cathode materials.For layered cathodes,the widelyemployed cationic dopants include K+[90],Na+[91,92],Mg2+[93,94],Al3+[95–97],Ti4+[98,99],Nb5+[100–102],Mo6+[103],W6+[104],and so on.Meanwhile,there are also various examples of successful modifications on the layered structures by anions [105],for example,F-and S2-.Generally,dopants regulate the cation mixing in the structureviathe following ways:1)enlarging the ionic radius difference between 3aand 3bsites;2)stabilising the structureviastrong dopant-O bonds;3)increasing the energy barrier of Ni migration from TM slab to Li slab;4) utilising the intense superexchange interactions.Here,the dopant has been categorised into monovalent,between bivalent and tetravalent,and higher than tetravalent,and the up-to-date doping strategies will be systematically reviewed.By detailing the corresponding working mechanisms of added dopants,an in-depth understanding of the role of elemental substitution towards cation mixing has been provided.
5.2.1.Monovalent cation doping
Alkali ions (e.g.,K+,Na+) can be introduced into the matrix of the layered cathode materials to occupy the Li site to improve the structural stability and regulate the cation mixing degree efficiently.For example,3% Li+was successfully substituted with Na+in the NCM523 material,which reduced the amount of anti-site Ni from 2.59 to 1.92%[91].As is shown in Fig.9a,the Na-doped NCM523 sample delivered an ultra-high rate capability (60.09 mAh g-1) at 50 C,compared with that of the bare-NCM523 (6.01 mAh g-1) [91].Similarly,the Li/Ni mixing reduction in the LiNi0.8Co0.15Al0.05O2upon Na doping was also observed,where the cycling stability was improved(Fig.9b)[92].Chen et al.found the K+doping can also reduce the Li/Ni mixing from 3.37 to 2.77% in NCM523 and brought about an initial charge capability of 210.3 mAh g-1(Fig.9c)and a capacity retention of 83.0%(Fig.9d) [90].

Fig.9.(a) Rate capability compared for NCM523 and Na-doped NCM523.Reproduced with permission from Ref.[91]Copyright The Royal Society of Chemistry 2014.(b)dQ/dV curves for Na-doped NCA.Reproduced with permission from Ref.[92].Copyright 2016,American Chemical Society.(c)Initial charge and discharge capacity and (d) Cycle stability for NCM523 and K-doped NCM523.Reproduced with permission from Ref.[90].Copyright 2019 the Authors.
The ionic radius of Na+and K+is larger than Li+,which contributes to the significant difference with that of Ni2+at the TM layer and expands the lattice space.Therefore,the Li/Ni mixing degree was suppressed in the modified samples [91].However,the Li layer doping could occupy the active sites during the electrochemical process,which suppress the battery capacity.Therefore,to achieve structure stability and minimise the influence on Li+diffusivity,the amount of dopants in the Li layer needs to be carefully controlled.
5.2.2.Between bivalent and tetravalent
Mg can be used as a dopant to stabilise the structure of the layered cathode material[33].For example,Sun et al.reported that Li/Ni mixing in NCM333 decreased from 2.1 to 1.4% by replacing TM ions by Mg2+.They attributed the reduction of cation mixing in the active material to the suppression of the superexchange effect and charge neutrality.As a result,initial discharge capacity increased from 164.8 to 176 mAh g-1[93].Moreover,the migration of Mg2+from the TM layer to Li layer in LiNi0.8Co0.2O2has also been noticed,which reduce the Li/Ni mixing by improving the interaction between Li slabs and O-M-O layer [106].It has also been noted that substituting Li+by Mg2+also reduce the Li/Ni mixing degree due to the difference in ionic radius between Mg2+and TM ions [107].Therefore,Mg doping efficiently suppress the cation mixing degree in the layered structure,with different doping sites attributing to different working principles.
Due to the high bonding energy of Al–O and the similar ionic radius to Ni3+,Al3+has been widely used as a low-cost and effective dopant to stabilise the structure and decrease Ni content to regulate the cation mixing in the Ni-rich cathodes.For example,Al partly replaced Mn to reduce the Li/Ni mixing degree in NCM811 and the capacity retained 78 mAh g-1after 1000 cycles at 10 C,significantly higher than the undoped sample(22 mAh g-1)(Fig.10a)[95].The replacement of Mn4+by Al3+leads to the transformation of Ni from bivalence to tri-valence;due to the charge neutrality,the reduction of the cation mixing has been achieved.

Fig.10.(a)Cycling performance for NCM811 and Aldoped NCM811 at 10 C.Reproduced with permission from Ref.[95].Copyright 2018 Elsevier Ltd.All rights reserved. (b) Reconstructed crystal structures of LiNi0.6Co0.15Al0.05Mn0.2O2 based on NPD refinement.Reproduced with permission from Ref.[96].Copyright 2019,American Chemical Society. (c) Cycle performance for the LiNi0.8Co0.15Al0.05O2 and LiNi0.7Co0.15Mn0.15O2 at 0.1 C.Reproduced with permission from Ref.[108].Copyright Wiley-VCH Verlag GmbH &Co.KGaA,Weinheim.
Additionally,Han et al.mentioned that Al doping at the Ni sites in NCM622 also achieve the reduction of the cation disordering(Fig.10b),where the modified sample showed the lowest cation mixing degree of 3.0% alongside an enhanced rate capability(104 mAh g-1at 10 C) and superior capacity retention (94.2% after 100 cycles at 0.2 C) [96].The direct substitution of Ni by Al could reduce the amount of Ni2+and the Li/Ni mixing degree in the layered cathode materials.Meanwhile,the similarity of the Ni2+and Li+ionic radius has been minimized,the cation mixing degree therefore reduced.
Moreover,the effect of Al3+substitution on preventing deterioration of Li/Ni mixing during electrochemical process has also been confirmed.The XRPD refinement results of the electrodes after 1500 cycles showed that the Li/Ni mixing of Al-substituted NCM increased from 1.8 to 9.1% in sharp contrast to that of bare-NCM (from 2.9 to 12.8%) [108].The strong Al–O bond stabilise the layered structure to prevent the TM migration during the cycle therefore reducing the cation mixing degree.As a result,the modified NCM delivered a higher capacity retention of 80% after 1500 cycles in the full batteries at 0.1 C (Fig.10c) compared with 75% of the bare-NCM[108].
It has been extensively reported that the introduction of Ti-doping can significantly influence the cation mixing degree and electrochemical performance of layered oxides cathodes [3].For instance,Ti-doped NCM811 (Fig.11a) material exhibited a reduced Li/Ni mixing from 4.4% to 2.5%,and it delivered an initial discharge capacity of 214.9 mAh g-1at 0.1 C alongside a columbic efficiency of 82.5%[99].Ti sufficiently suppress the formation of the O vacancies and prohibit the Ni migration during cycling,thus leading to the reduced amount of Li/Ni mixing during electrochemical cycling (Fig.11b) [99].Moreover,Markus et al.employed density functional theory (DFT) calculations to confirm the effects of Ti doping on suppressing the undesired phase transformation during charge/discharge (Fig.11c).The free energy of the formation of the rock-salt phase has been increased after Ti4+is doped [98].Meanwhile,due to the higher O-binding energy of Ti than other TM elements,the frame structure was stabled by Ti doping,the migration of the TM ions and the formation of the vacancy has been restricted[109].Ti was also employed in the high-voltage LCO layered cathode in which Ti was found to segregate at the surface of the primary particles [110].As the hazardable phase transformation usually happens at the particle surface and propagates to the material bulk,the enhanced surface stability of the modified samples by surface-segregated Ti prevents the occurrence of this scenario from its origin.

Fig.11.(a) Comparison of crystal structure for pristine and Ti-modified NCM811. (b) XRD diffraction pattern for cycled NCM811 and Ti-doped NCM811.Reproduced with permission from Ref.[99].Copyright 2019 Elsevier Ltd. (c) Calculated free energy of formation at room temperature for rock salt structure from NCM structures with different lithium concentration.Black-colour line (Ti00) corresponds to unsubstituted NCM,and the red(Ti03)corresponds to Ti-substituted NCM811.Reproduced with permission from Ref.[98].Copyright 2014 American Chemical Society.
The dopants between bivalent and tetravalent could facilitate a stable structure due to the stronger dopant-O bonds,which prohibit the TM migration.Although substitution elements are various,doping sites are usually definite and unitary.The possibility of multi-sites co-doping into the layered cathodes is worthy to be explored.It may realise the proper cation mixing degree and enhanced electrochemical performance of layered cathodes;meanwhile,enrich the design of better cathode materials.
5.2.3.Higher than tetravalent
Taking advantage of the large ionic radius and strong Nb–O bond,Nb expand the lattice parameters and stabilise the material structure of the layered oxides cathodes materials.However,the influence of Nb on cation mixing remains debating.Li et al.claimed that 2% Nb5+doping in NCM811 has reduced the Li/Ni mixing degree from 1.78% to 1.66% and improve capacity retention and discharge capacity of the active material(Fig.12a) [100].Similarly,the Nb doping in the NCM622 also realised the reduction of the reduction of the cation mixing and the improved cycle stability(Fig.12b)[101].The decreased cation mixing degree has been summarized as the strong Nb–O bond,which stabilise the host structure to prevent the TM migration and the formation of vacancy.
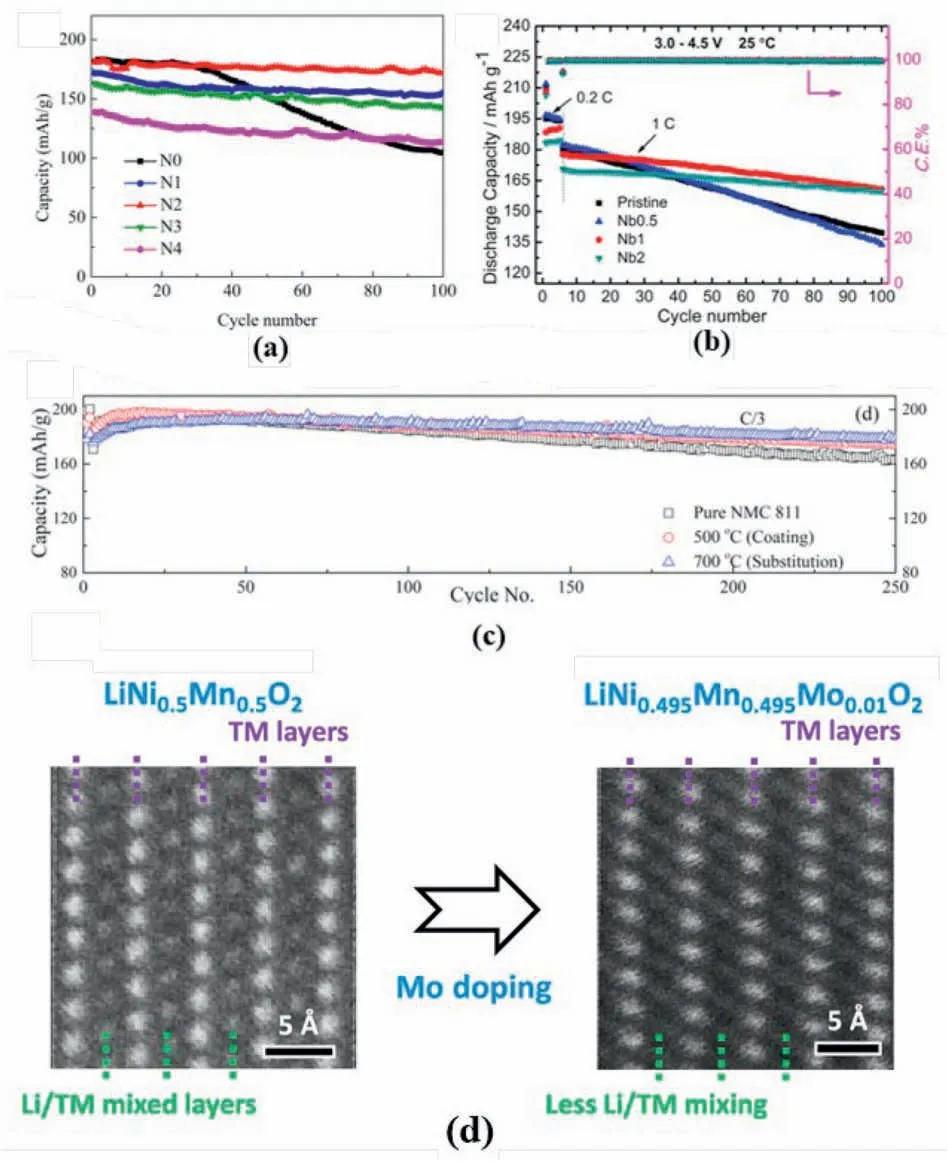
Fig.12.(a) Cycle performance of Nb5+ modified NCM811 and bare-NCM811.Reproduced with permission from Ref.[100].Copyright 2020 Elsevier B.V. (b) Cycle performance for Nb-doped NCM622 and bare-NCM622.Reproduced with permission from Ref.[101].Copyright 2017 Elsevier B.V. (c) Cycle stability for Nb-modified NCM811 under differing sintering temperature.Reproduced with permission from Ref.[102].Copyright 2021 American Chemical Society.(d) HAADF STEM images of the pristine and Mo-doped LiNi0.5Mn0.5 to illustrate the cation mixing.Reproduced with permission from Ref.[111].Copyright 2019 American Chemical Society.
However,Xin et al.had a different finding that Nb doping increased Li/Ni mixing due to the high valence of Nb together with the formation of Ni2+[102].Although the Li/Ni mixing degree in NCM811 increased,as shown in Fig.12c,enhanced capacity retention has been observed(93.2% after 250 cycles at 1/3 C) [102],which confirmed the positive effect of cation mixing in the structural stabilisation of layered cathodes.The authors have allocated the charge balance after Nb5+was doped to the host structure to the increase of the cation mixing degree.
Other high-valance state elements,such as Mo6+and W6+,were also introduced into the layered cathodes to regulate the cation mixing degree.For instance,comparing with the bare sample,the Mo6+doped LiNi0.5Mn0.5O2delivered a reduced Li/Ni mixing degree as revealed by the STEM-HAADF image(Fig.12d).As indicated by DFT calculation results,replaced Ni2+by Mo6+,the structure with less antisite defects is more stable.The formation energy of the structure without cation mixing is-6.24eV,significantly lower than the one with anti-site defects[111].Meanwhile,after Mo6+doping,the localized charge environment in the material is altered,this has also been attributed to the reduction of the cation mixing [111].By contrast,after Mo6+was doped into NCM523,the increase in cation mixing degree has also been noticed.As supported by the XPS results (Fig.13a),the increased Li/Ni mixing degree was attributed to the increase of Ni2+content due to charge balance after heterovalent substitution[103].Additionally,Chu et al.introduced W6+into the host structure to enhance both battery capability (197.8–201.6 mAh g-1at 0.2 C) (Fig.13b) and cycle stability (90.6–96.7% at 1 C)(Fig.13c)[104],in which an increased cation mixing behaviour was also observed after W-substitution.The increase in the cation mixing has been attributed to the increased Ni2+amount,which was generated by charge balance.

Fig.13.(a)XPS data for Mo-modified samples for as-prepared cathode materials before and after 500 cycles from 3.0 to 4.3 V,and 4.6 V.Reproduced with permission from Ref.[103].Copyright 2017 Elsevier B.V. (b) charge-discharge profiles at 1st cycle at 0.2 C. (c) cycling stability over 3.0–4.5 V at 25 °C.Reproduced with permission from Ref.[104].Copyright 2021 American Chemical Society.
Generally,high-valance dopant substitution in the material structure leads to more Ni2+as a result of charge balance,which increases the cation mixing degree.However,the high valent dopants could alter local charge environments,which influence cation mixing differently at local level [108].Therefore,a homogeneous distribution of the doping elements in the host structure benefits the consistency of local level and electrode level cation mixing degree.Moreover,high valent cations,such as Nb5+,Mo6+,have larger ionic radius,which may distort the lattice structure of the host material.This lattice distortion needs to be carefully considered and worthy to be uncovered whether it influence the cation mixing.
5.2.4.Anion doping
Anion doping has been employed to regulate the cation mixing of the layered structure.For example,Li et al.introduced the F-into LiNi0.85Mn0.075Co0.075O2,which,although reducing the initial discharge capacity,achieves a high capacity retention of 94 and 83.3% at 1 C after 200 cycles at RT and 55°C,respectively (Fig.14b and c) [105].The enhancement of cycle stability could be attributed to the increase of the cation mixing degree in the host structure.As shown in Fig.14a,all the formation energy of the superexchange interaction decreased after halogens doping.Especially for the F-,the dramatically reduced formation energy of the antisite defect indicated the increase of the cation mixing degree.To study the mechanisms of anion doping,the effects of widely used F-,Cl-and S2-have been studied by density functional theory(DFT) calculations in tuning the Li/Ni mixing of LiNiO2[112].The results indicated that different anions had various influences on cation mixing,in which S2-reduces the cation mixing degree,while F-and Clpromote the formation of Li/TM antisite defects.The F-and Cl-doping could intensify the superexchange interaction in the structure;however,S2-reduced the occurrence of the superexchange interactions compared with O2-[112].
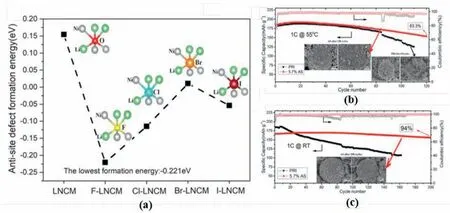
Fig.14.(a)Effect of F,Cl,Br and I substitution on calculated antisite defect formation energy of the inset model.Cycle performance for F-doped and bare samples at(b) 55 °C and (c) Room temperature.Reproduced with permission from Ref.[105].Copyright 2018 the Authors.Published by Wiley-VCH Verlag GmbH &Co.KGaA,Weinheim.
Compared with cation doping,less efforts have been made on the anion substitution to control cation mixing in the layered oxides.In addition to the single anion doping (F-,Cl-,S2-),it has been reported that the polyanionic groups,such as P[113]and B[114],could also benefit the electrochemical performance of the layered cathodes.However,its influence on cation mixing remains unclear,where more attention needs to be paid on the investigation of its underlying mechanisms.It is also noted that anions are difficult to be characterised with current technologies.Accordingly,more advanced characterisation tools are needed to build the fundamental chemistry/structure/function relations within anion-doped layered oxides.
5.3.Surface engineering
In addition to optimising calcination parameters and elemental substitution strategies,surface engineering also effectively impacts the cation mixing degree of the layered cathode materials.The TM migration in the layered cathode materials induce the undesired phase transformation of active materials,which usually occurs at the surface region of material particles and propagates to the material bulk during electrochemical reactions.Different coating materials,such as metal/nonmetal oxides and polyanion,effectively prevent the phase evolution generated at surface region from propagating to the bulk region,thereby improving the cycle stability and battery capacity of the layered cathode materials.
For instance,Feng et al.coated the NCM811 with Al2O3and found that the Li/Ni mixing in the layered structure can be suppressed by 19%.The initial charge capacity increases from 201.1 to 207.9 mAh g-1,together with an improved capacity retention of 82.67% after 100 cycles at 0.5 C[115].However,it is also found that a 7% ZrO2coating increased the Li/Ni mixing degree in LiNi0.82Co0.09Mn0.09O2(NCM82),which benefits the capacity retention (Fig.15a),but the detriment on battery capacity has also been noted[116].Moreover,it has been reported that LiFePO4surface modification on NCM622 sufficiently regulate the cation mixing in the Ni-rich cathode materials.After the LiFePO4coating,the capacity retention of the modified sample was successfully improved to 86.5% in a full cell battery for 1000 cycles under 4.6 V [117].It is indicated by the first-principal calculation that the total energy for Pto absorb on oxygen sites,TM sites and Li sites along the (1 0 4) are different (Fig.15b).The TM and lithium sites deliver the lowest absorption energy(-817.66 eV),which reveals the TM sites are preferable for the anionic group to absorb[117].Furthermore,due to the presence of the LiFePO4layer,the surface oxygen evolution at the delithiation stage has also been suppressed to prevent the TM ions migration at the surface region.
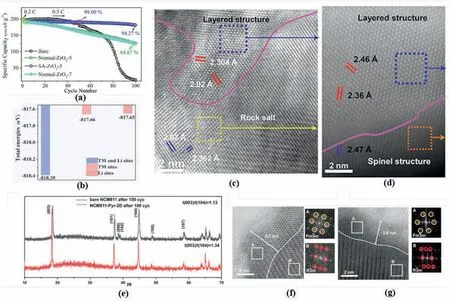
Fig.15.(a) Cycle performance of Zr2O3 surface modified NCM82.Adapted from Ref.[116].Copyright 2022 Elsevier BV (b) Total energy comparisons of three possible sites for PO43-adsorption.Reproduced with permission from Ref.[117].Copyright 2019 American Chemical Society.HADDF-STEM images of the(c)pristine NCM333 and(d)AlPO4 coated NCM333 after 100 cycles.Reproduced with permission from Ref.[119].Copyright 2019 American Chemical Society.(e)XRPD patterns of the bare NCM811 and NCM811-Pyr-2D after 100 cycles.STEM images of the(f)pristine NCM333 and(g)AlPO4 coated NCM333 after 100 cycles.Reproduced with permission from Ref.[120].Copyright 2019 American Chemical Society.
Apart from metal/non-metal oxides and polyanion,other coating materials,such as sulphate-based surfactant [118],organic complex[119],have been proven to be helpful in restricting the cation mixing of layered cathode materials.For example,a metal phosphate organic complex has been reported to coat NCM333,which delivered nearly unchanged antisite defects (2.82%–2.83%) compared with the pristine sample.However,the rock-salt structure at the surface has been observed in the 100 times cycled pristine sample in Fig.15c,by contrast,only a 5 nm spinel like structure was observed in the AlPO4coated sample in Fig.15d [119].It has been confirmed that,the antisite defect has been prevented by the coating materials to propagate in the bulk region.Additionally,Jerng et al.coated the two-dimensional (2D)pyrazine-linked covalent organic framework (Pyr-2D) on the NCM811.The 100 times cycle stability of the surface modified NCM811 was increased from 17.9% to 68.2% under 1 C between 2.7 and 4.5V.As illustrated in Fig.15e,the ratio of I (0 0 3)/I (1 0 4) indicated that the Pyr-2D coated NCM811 delivered the lower cation mixing degree after 100 cycles [120].Meanwhile,comparing with the bare NCM811,a smaller antisite defect and spinel-or rock-salt-like phase(3.6 vs 5.1 nm)was detected in the NCM811-Pyr-2D as shown in Fig.15f and g[120].
The results confirmed surface modification successfully prevent cation mixing from intensifying and undesired phase formation.It is accepted that TM migration and dissolution at the surface region induce structural instability and further trigger bulk destruction.The surface modification help build a stable surface to prevent the TM migrationinduced cation mixing and undesirable phase transformation propagating to the bulk region.Besides,it is strongly recommended to compare the pristine and cycled electrodes when studying the improvement mechanism on the surface modification.The combination of STEM and XRPD characterisation on the cycled electrodes could confirm whether the surface modification sufficiently prevent the TM/Li mixing at the surface region.Further,the ions from the coating materials could dissolve into the host structure,unavoidably forming a modified sub-surface.Therefore,ion diffusivity and structure stability of the sub-surface need to be considered when selecting the coating materials.
6.Conclusion and future perspective
TM/Li mixing is essential to the electrochemical performance of layered oxides cathode materials used in LIB.Origins for cation mixing include:1)a similar ionic radius with Li+and Ni2+,2)reduced entropy of the system,3) reduced magnetic frustration,4) superexchange interactions,and 5)migration of TM ions.The similarity in ionic radius of Ni2+and Li+is commonly used to explain the origins for Ni/Li mixing in the layered oxides,whereas TM migration is used to explain Co/Li or Mn/Li disordering.Additionally,system entropy reduction,reduced magnetic frustration and superexchange interactions should be considered regarding the formation of the cation mixing.
Cation mixing significantly reduces Li+diffusion efficiency,charge/discharge capacity,initial columbic efficiency,and rate capability of the active materials.However,cation mixing stabilises the material structureviapillar effect and significant superexchange interactions.The degree of cation mixing in the material structure might be optimisable for capacity retention and minimal charge/discharge capacity reduction,initial columbic efficiency,and rate capability of layered oxides.However,the variety of the layered oxides,including synthesis and different elemental composition,and limitations of available characterisation techniques,make this practically difficult.
Detection of cation mixing in the structure is dependent on advanced techniques including X-ray/neutron powder diffraction,electron microscopy,NMR and PDF.Localised(1–5 nm)and electrode-level(2–10 μm)information on cation mixing,can be readily evidenced;however,at the transitional particle-level(~1 μm)cation mixing is largely indeterminate at present.Importantly,therefore,there can be significant differences in findings from localised and electrode-level mixing in the material.This was evidenced when,for example,X-ray phase contrast holotomography was used in a study of NCM811 cathodes during electrochemical cycling and asynchronous behaviour of particles was found that corresponded to inconsistency/incoordination in electrochemical reactions in different particles [121].Asynchronous behaviour occurs also between oxides particles during charge/discharge.X-ray phase contrast holotomography might therefore be developed to quantitatively assess connections between localised and mean,cation mixing.
Cation mixing in the layered oxides cathodes is impacted by multifactors,including,sintering temperature and atmosphere and cooling rate.Generally,reasonably low sintering temperature and elongated holding-time,together with O2atmosphere result in low cation mixing being exhibited in the active material.Elemental substitution can be used to regulate cation mixing by minimising the similarity in ionic radius,reducing Ni2+numbers and restricting migration of TM ions.Additional understanding is needed of the modification sites of the dopants in the structure in which substitution at the Li site minimizes influence on transportation of Li ions,whilst that at TM sites,prioritises restriction of TM migration during charge/discharge.
Surface engineering is used to stabilise the surface/subsurface of the layer-structured materials,thereby diminishing degree of cation mixing in the crystal structure.However,the impact of interfacial compatibility between coating layers and bulk material and ionic/electronic conductivities of the coating layer,will need to be confirmed before surface modifications.Interestingly,recent studies show that electrolyte modification might be practically used to control cation mixing of electrode materials.An example isN-allyl-N,N-bis(trimethylsilyl)amine (NNB) as an electrolyte additive in batteries containing LiNi0.8Co0.15Al0.05O2and Li electrodes.This contributes to the formation of NNB-derived Si-containing SEI films that regulate Li/Ni disordering during cycling[122].
Here,we have systematically shown the disadvantage of cation mixing in the prevalent layered oxides cathodes for LIBs towards restricting Li+diffusion and formation of undesirable phase evolution,where the initial columbic efficiency,discharge capacity and rate capability can be relatively reduced;however,the advantages of cation mixing regarding the pillar effects and superexchange interaction to stabilise the structure has also been noted,where the capacity retention can be improved.Tunning the calcination conditions,exploring the multi-sites elements substitution and developing a stable electrode/electrolyte surface minimise side effects and maximum benefits of the cation mixing degree.Therefore,we propose to attach importance to the double-edged effects of the cation mixing when developing high energy layered cathode materials.
Declaration of competing interest
Zaiping Guo is an editorial board member for [Nano Materials Science]and was not involved in the editorial review or the decision to publish this article.All authors declare that there are no competing interests.
Acknowledgements
J.X.Li acknowledges the University of Adelaide for providing scholarship to support the Doctor of Philosophy candidature.Dr G.M.Liang acknowledges the Australian Institute of Nuclear Science and Engineering(AINSE)Limited for providing financial assistance in the form of a Post Graduate Research Award(PGRA)to carry out this work.This work is supported by the Australian Research Council under grants DP200101862,DP210101486,and FL210100050.
杂志排行

Namo Materials Science的其它文章
- MXene-based flexible pressure sensor with piezoresistive properties significantly enhanced by atomic layer infiltration
- Construction of all-organic low dielectric polyimide hybrids via synergistic effect between covalent organic framework and cross-linking structure
- Synergistic coupling of 0D–2D heterostructure from ZnO and Ti3C2Tx MXene-derived TiO2 for boosted NO2 detection at room temperature
- Wearable and stretchable conductive polymer composites for strain sensors:How to design a superior one?
- An overview of recent progress in the development of flexible electrochromic devices
- Water-based synthesis of nanoscale hierarchical metal-organic frameworks:Boosting adsorption and catalytic performance