少毛症-淋巴水肿-毛细血管扩张-肾缺陷综合征*
2024-01-22王文红
刘 涛 王文红
天津市儿童医院(天津大学儿童医院)、天津市儿童出生缺陷防治重点实验室肾脏科(天津 300134)
少毛症-淋巴水肿-毛细血管扩张-肾缺陷综合(hypotrichosis-lymphedema-telangiectasia-renal defect syndrome,HLTRS)又称少毛症-淋巴水肿-毛细血管扩张-膜增生性肾小球肾炎综合征,是由SOX18基因突变导致的以少毛症、淋巴水肿、毛细血管扩张及补体正常的膜增生性肾小球肾炎为主要临床特征的一种罕见遗传性疾病。HLTS的患病率不足百万分之一,目前国外只报道11例SOX18基因变异所致的HLTS患者,其中伴有肾损害的只有3例,2例肾损害表现为膜增生性肾小球肾炎[1],1例表现为蛋白尿(未行肾穿刺病理检查)[2]。另有2例未行基因检测的病例报道,临床表现均为少毛症、毛细血管扩张,同时伴有膜增生性肾小球肾炎[3-4]。虽国内尚无HLTRS的相关病例报道,但可能存在对该病认识不足而导致HLTRS在国内的发病率被低估。HLTRS尚无明确有效的治疗方法,其严重程度与SOX18基因突变的位点相关,严重胎儿期即死亡或发病,而目前报道有幸顺利出生的患儿,虽可以达到长期存活,但多伴有严重症状性高血压,并可进展至终末期肾病,需要肾脏替代治疗或肾移植治疗。本文主要对该病的发病机制及诊治进展进行综述。
1 病因及发病机制
HLTRS与SOX18基因变异有关,为常染色体显性遗传。SOX18编码一种转录因子,常表达于生长中的脉管系统,参与血管、淋巴管、毛囊的发育过程,SOX18基因突变可引起这些系统不同程度的发育受损,从而导致HLTRS的发生[5]。
1.1 SOX18基因变异SOX18是自发性基因突变小鼠品系Ragged的人类同源物,SOX18有四个不同的突变体,分别为Ra、RaJ、Ragl和RaOp[6]。SOX18的杂合子(显性)或纯合子(隐性)突变最初出现在HLTS家族中,之后逐渐发现SOX18基因的杂合子突变(如c.720 C>A)与HTLRS相关。SOX18由一个DNA结合域(HMG盒)和两个反式激活域(TAD)组成,一个TAD紧邻HMG盒的下游(被称为中央TAD),另一个TAD是在C端结构域的9个氨基酸反式激活基序(被称为9aa TAD)(见图1)[6]。HLTS除一例患者为HMG 框最后一个氨基酸的无义突变,其余报道病例均是由位于 HMG 盒中的错义突变(常染色体隐性遗传)或中央 TAD中的无义突变(常染色体显性遗传)引起的[2,5]。直至目前,报道的HLTRS均为中央TAD的无义突变引起的,遗传方式为常染色体显性遗传。
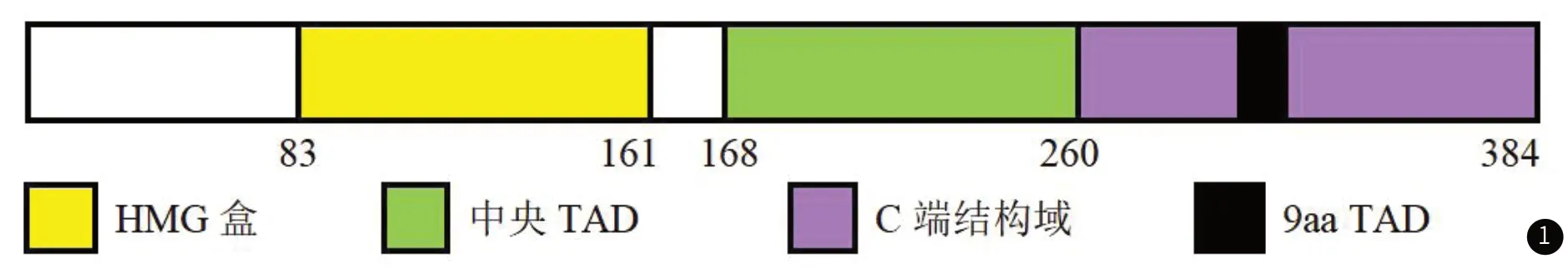
图1 SOX18蛋白组成示意图
1.2 显性负性SOX18突变蛋白的遗传分子机制小鼠中的SOX18显性突变可合成反式激活功能受损的截短SOX18蛋白[7],相应的表型以皮毛缺陷、心血管异常以及淋巴缺陷为特征[6]。目前报道的人类SOX18显性负性突变会导致罕见的HLTRS,患者表现出明显的毛囊缺陷(如毛发稀疏或缺失)、淋巴缺陷(如淋巴管渗漏引起的四肢肿胀)以及血管缺陷(包括毛细血管扩张症及导致肾衰竭的血管缺陷)[8]。Alex J McCann 等[8]利用SOX18的显性负性突变体SOX18RaOp对SOX18的显性负性机制进行了研究,发现SOX18RaOp突变蛋白主要通过以下几方面产生致病作用:①SOX18RaOp突变蛋白是一种有效的转录抑制因子。这种截短的蛋白质仍然能够与DNA结合,但不能激活基因转录,因此可将SOX18蛋白质的功能降低至25%以下,且SOX18RaOp与染色质的结合稳定性增加,使其在细胞核中积累,增加了转录因子核内浓度,放大了其有害作用。SOX18的显性负性突变体为纯合子时是胚胎致死性的,只有杂合子才能存活,在杂合子的情况下,SOX18与SOX18RaOp同时存在于细胞中,但增加野生型SOX18的剂量并不能抑制SOX18RaOp突变蛋白引起的转录活性缺乏。②SOX18RaOp突变蛋白在全基因组范围内干扰SOX18的结合位置。SOX18RaOp与染色质的结合力更高,不仅具有抑制原本活跃的基因组区域的潜力,且使SOX18与染色质的结合数量急剧减少,并可将SOX18募集到基因组的转录沉默区域。③SOX18RaOp突变蛋白干扰SOX-F家族成员的转录活性。SOX18RaOp可以招募SOX7和SOX17,从而抑制SOX7和SOX17的作用,这种机制可以解释为什么SOX18RaOp突变小鼠会出现严重的血管缺陷,而SOX18基因敲除的小鼠却没有心血管缺陷。④SOX18RaOp突变蛋白扰乱了SOX18的寡聚状态。SOX18蛋白的一个功能特征是其形成同源二聚体的能力,这是一种与内皮细胞特异性转录特征密切相关的分子状态。SOX18RaOp可能竞争性结合SOX18,干扰SOX18同源二聚体的产生。5.SOX18RaOp干扰SOX18同源二聚体的特异性蛋白伴侣MEF2C与染色质的结合。MEF2C对血管发育至关重要,其表达显著增强了SOX18的转录能力,而SOX18RaOp无法使MEF2C稳定在染色质位点上。显性负性SOX18突变蛋白不仅使SOX18转录功能下降,且其保留了结合蛋白伴侣及招募SOX-F家族的能力,这可能会干扰多个调节,从而使负面作用放大,因此不同的显性突变位点与疾病表型严重程度相关,突变蛋白越长,临床表型越严重,而随着突变蛋白变短,它会失去对其他蛋白质复合物的干扰能力,临床表现就相对较轻。尽管目前研究在显性负性SOX18突变蛋白的分子机制方面取得了相当大的进展,但这些变化如何与人类和小鼠中观察到的突变表型直接相关仍然未知。
1.3 SOX18基因突变与肾缺陷的关系显性负性SOX18突变蛋白可以通过抑制SOX18的转录功能而影响肾脏血管及淋巴管发育,可导致与血栓性微血管病相似的MPGN[1]。研究发现,小鼠Sox-/-突变仅表现出轻度皮毛缺陷[9],而Sox17/Sox18双杂合子突变小鼠可见肾脏改变[10],人类SOX17基因突变与先天性肾脏和泌尿道畸形(CAKUT)如膀胱输尿管反流、肾盂输尿管连接部梗阻相关[11]。显性负性SOX18突变蛋白除可以通过抑制SOX18的转录功能而影响肾脏血管及淋巴管发育外,还可以通过招募SOX17而抑制SOX17的作用导致肾脏及泌尿道的异常。SOX18也可能通过与其他基因的相互作用在肾脏血管发育中发挥作用。例如,基质金属蛋白酶 7(MMP7) 在先天性肾发育不良患者中过表达[12],临床特征是正常肾发育受阻、囊肿形成、肾生长受损以及肾单位减少或缺失。MMP7抑制某些受骨形态发生蛋白7(BMP7)刺激的细胞中分支结构的形成,而BMP7对正常的肾脏发育至关重要。 而MMP7是SOX18的靶标,并与SOX18共同表达于人体皮肤的血管中[11]。当SOX18基因发生突变后,也会干扰MMP7的作用,从而引起CAKUT的发生。
2 临床表现
2.1 HLTS相关临床表现目前国外报道共11例SOX18基因突变相关的HLTS(其中包括2例HLTRS),临床表现除少毛症、淋巴水肿及毛细血管扩张外,还存在多脏器受累、特殊面容等异常表现[2,13-14]。⑴少毛症:所有SOX18突变的患者均没有眉毛和睫毛,头皮毛发稀疏,或完全脱发,症状出现早,多在生后6月内出现。其次随着年龄增大,会发现阴毛及腋毛缺如。⑵淋巴水肿:①胎儿期:可有腹水、心包积液、胸腔积液、乳糜胸、乳糜性腹水、肾积水、肠管扩张、羊水过多、胎儿水肿、胎儿窘迫、死胎、早产、过期产。②出生后:最常见为下肢淋巴水肿,发病年龄差异很大,从出生至15岁不等。其他可见的水肿部位为面部、眼睑、鞘膜积液、全身皮肤水肿、浆膜腔积液(心包积液、胸腔积液或腹水)。需要注意的是,并不是每位患者均会出现淋巴水肿的症状,即使出现了该症状,亦不是持续存在,可能会间断出现。⑶血管缺陷:①毛细血管扩张:表现为红色或紫红色丝状、点状、星芒状或片状红斑,最常见部位为手掌,其次为足底、头皮、阴囊、下肢等部位。亦可表现为皮肤小的丘疹性血管病变、湿疹、血管瘤、血管痣、大理石样皮肤、网状青斑、静脉曲张或皮肤异色病。②其他血管缺陷表现:肺动脉高压、主动脉扩张、鞘膜积液、频繁鼻出血。⑷其他系统异常表现:HLTS还可以引起其他各器官系统的病变,包括①颅面部畸形:短头畸形,鼻梁变宽,唇厚,牙龈肥厚,凸颌;②神经系统:可有学习障碍、阅读障碍、头痛、脑钙化表现;③头颈部:可有先天性斜颈;④心血管系统:可有高血压、升主动脉扩张、主动脉根部扩张、肺动脉高压、双向动脉导管未闭;⑤呼吸系统:可有出生时呼吸窘迫综合征、婴儿期肺水肿、新生儿乳糜胸、间质性肺疾病;⑥消化系统:可有肠套叠、无症状类癌肿瘤、可复性腹股沟疝、先天性回肠闭锁;⑦泌尿系统:可有肾衰竭、膜增生性肾小球肾炎、蛋白尿、血尿;⑧内分泌系统:可有身材矮小、骨龄延迟;⑨皮肤及附属器官:可有皮肤薄而透明,趾甲发育不全,下肢皮肤硬化,脐带短,汗多,伤口愈合不良,皮肤损伤后的萎缩性瘢痕;⑩癌症:可有基底细胞癌[1]。
2.2 补体正常的膜增生性肾小球肾炎目前报道的4例HLTRS肾组织病理均显示膜增生性肾小球肾炎(MPGN),其中3例明确为I型MPGN[3-4,15],1例未描述MPGN的病理分型[2]。4例HLTRS临床上可表现为肾炎(1/4)、肾病综合征(1/4)及肾衰竭(2/4)。本病几乎均存在血尿及蛋白尿,其中1例可见发作性肉眼血尿,其余为持续镜下血尿,蛋白尿可表现为轻度至大量蛋白尿不等。表现为肾病综合征的患儿亦伴有高脂血症、低白蛋白血症、正细胞正色素性贫血。3例HLTRS出现严重高血压。3例确诊HLTRS时有血肌酐升高,其中2例在确诊后5~8年出现肾衰竭,并接受肾移植治疗,移植肾在随诊过程中功能维持正常。
2.3 总结既往HLTRS病例的临床表现目前国内尚无HLTRS的相关报道,国外共报道4例确诊为HLTRS的病例,其中2例经基因检测为SOX18基因显性突变,另2例未行基因检测。4例患儿行肾活检均提示MPGN。Richard等[2]还报道1例SOX18基因显性突变的HLTS伴有蛋白尿,但未行肾穿刺病理检查。表1总结了4例确诊为HLTRS病例的临床资料。
表1 4例LTRS病例的临床资料总结

表1 4例LTRS病例的临床资料总结
注:“-”表示文献中未描述。
病例1 病例2 病例3 病例4参考文献 Sherwood MC, et al [1987] [1999] Irrthum, et al [2003] Proesmans W, et al [1989]Moalem S , et al [2015] Moalem S, et al [2015]性别 男 女 男 男诊断时年龄 4岁 10.5岁 6月龄 10岁家族史 有/无 有 无 有成员及表现 患儿父亲终末期肾病、高血压、- 哥哥于胎龄3周时宫内死亡,尸 祖母、姐姐均表现为少毛症(头发稀疏,皮肤异色病,肾脏病理为MPGN 检显示胎儿水肿,心包、胸腔、 眉毛及睫毛缺如)、高血压或局灶节段性肾小球硬化 腹腔乳糜性积液,全身血管充血伴肺淋巴管扩张SOX18基因突变-- c.720C>A p.Cys240* 杂合子 c.720C>A p.Cys240* 杂合子遗传方式 常显?- 常显 常显种族(国籍/人种) 英国/白种人 加拿大/白种人-/白种人 比利时/白种人产前B超-- 无特殊 无特殊分娩方式 孕周 足月---方式 剖宫产---原因----少毛症 头发 稀疏 发际线后移 质地正常 稀疏 发际线后移 非常稀疏 非常稀疏眉毛 短 缺如 缺如 缺如睫毛 短 缺如 缺如 缺如阴毛、腋毛- 缺如--发现时间--6月龄 出生时淋巴功能障碍面部水肿-- +(婴儿期发现) +(出生时发现)眼睑水肿--- +(出生时发现)下肢水肿- + +(出生时发现)-鞘膜积液 无- +(婴儿期发现) +(出生时发现)皮肤 细血管扩张 +(面颊、手指伸侧、肘部伸侧) +(手指及脚趾伸侧) +(头皮、阴囊、下肢) +(双手伸侧、膝盖、肘部、鼻、牙龈)其他-- 面部湿疹 面部雀斑皮肤附属器官 指甲正常 指趾甲正常--面像 面色苍白 内眦赘皮 眼距宽 凸颌 面色苍白 眼距宽凸颌 嘴唇厚 凸颌 鼻根及鼻尖宽 嘴唇厚 凸颌 鼻根及鼻尖宽 鼻子狭长神经系统--- 学习困难 脑脉络丛钙化 右侧海马硬化头颈部 扁桃体、腺样体切除 听力检查正常 频繁鼻出血 眼科检查正常 频繁鼻出血 长期频繁鼻出血 面部基底细胞癌心血管系统 血压正常 高血压 5岁时行房缺修补术 高血压 高血压呼吸系统-- 肺水肿(婴儿期)-消化系统 幽门梗阻(1月龄)-- 肠套叠泌尿系统 临床表现 肾炎表现 肾病综合征表现 肾衰竭(5岁) 肾移植(14岁) 肾衰竭(18岁) 肾移植(27岁)血尿 2次粉红色尿;镜下血尿 镜下血尿(4~6个/HP)- 镜下血尿(0~145/μl)(9~ 50个/HP)蛋白尿 轻度蛋白尿(定性-~3+,半定量 大量蛋白尿(2.4g/24h)- 中度至大量蛋白尿[45~75 mg/(kg·24 h)]0.5~1.99)肾功能 血肌酐持续轻微升高(55~96μm 血肌酐升高(84μmol/l)- 血肌酐正常;肾小球滤过率90~112ml/ol/l);肾小球滤过率140ml/(min 肾小球滤过率55.8ml/ (min·1.73m2)·1.73m2) (min·1.73m2)血压 正常 严重高血压(212/124 严重高血压(207/155mmHg) 高血压mmHg)补体 C3、C4、CH50均正常 C3、C4、CH50均正常- C3、C4、CH50、C3D、C1q均正常肾脏B超 正常 大小正常,肾实质回声- 肾动脉硬化轻微增强肾脏病理 Ⅰ型MPGN Ⅰ型MPGN 慢性肾 MPGN 慢性微血管病变累及肾小 Ⅰ型MPGN 微血管病变血管病变 球及肾小球外血管其他化验 凝血功能、免疫球蛋白(Ig)、 血白蛋白28g/l;高脂血- 血清胆固醇、甘油三酯长期升高;类风自身免疫抗体、乙肝五项、 症(胆固醇10.2mmol /l); 湿因子、免疫球蛋白、自身免疫抗体、ASO 、染色体均正常 正细胞正色素性贫血(血 染色体均正常红蛋白81g/l);IgA、IgM正常,IgG降低;凝血功能、自身免疫抗体、乙肝五项、ASO、血小板、血友病因子、染色体均正常发育 生长发育正常(身高位于第25百 生长轻度发育迟缓(身高- 生长轻度发育迟缓(身高和体重均在第3分位,体重位于第50百分位) 131cm位于第10~25百分 百分位);皮下脂肪发育不良;精神运动位,体重31.9kg位于第 发育迟缓25~50百分位)其他 几次不明原因发热 容易擦伤- 过早衰老;容易擦伤
3 诊 断
因该病罕见,目前无明确的诊断标准,通过总结文献报道,临床上出现以下线索时应怀疑HLTRS:1.少毛症;2.淋巴水肿;3.毛细血管扩张;4.伴有肾损害如血尿、蛋白尿、肾功能异常等。对于临床疑似病例,需行肾穿刺病理检查及基因检测,若肾活检符合补体正常的MPGN或存在SOX18基因突变,并除外其他引起MPGN的继发因素,则可考虑诊断HLTRS。
4 治 疗
关于HLTRS的治疗,目前尚无指导方针及较多的临床经验,通过查阅总结文献发现HLTRS无有效的治疗方案,主要是对症治疗。
4.1 SOX18转录因子抑制剂普萘洛尔是非选择性β-肾上腺素能阻滞剂,是婴幼儿血管瘤的一线治疗药物。动物实验发现普萘洛尔以剂量依赖性方式恢复野生型SOX18的功能,其对野生型SOX18同源二聚体形成的抑制作用较温和,但对非功能SOX18/RaOp蛋白复合物的组装的抑制作用更强,普萘洛尔的R(+)对映异构体是一种有效的SOX18活性选择性抑制剂[16]。Jeroen Overman等[16]提出存在SOX18显性突变的HLTS病例[14,17]未发生肾脏损害可能是因为应用普萘洛尔治疗减轻了患儿的临床症状有关。对HLTS伴有心包积液患儿,应用布洛芬及泼尼松抗炎治疗无好转,改为普萘洛尔治疗,普萘洛尔起始量从0.8mg/(kg·d)分3次口服,每2~3周逐渐增加,直至达到4.1mg/(kg·d),在普萘洛尔增加至3mg/(kg·d)时心包积液开始消退,口服53周后心包积液完全消失[16]。这些基础实验及临床治疗经验可以看到普萘洛尔对HLTRS的治疗潜力,但仍然需要进一步观察。
4.2 关于MPGN治疗⑴糖皮质激素 Gupta IR等[4]报道先证者表现为肾病综合征型,给予甲强龙10mg/(kg·d),共3天,后改为泼尼松2mg/(kg·d),每日分2次进行治疗。后泼尼松逐渐减量,减量方式未描述。随诊2年后口服泼尼松20mg qod,血清白蛋白水平正常,尿蛋白持续存在,但由最初的2.4g/d下降至0.9g/d,存在持续高脂血症,肾功能未得到明显改善,血肌酐从最初84μmol/l上升至149μmol/l。⑵控制血压 建议参照儿童青少年高血压管理临床实践指南[18-19]。⑶其他 对于存在持续高脂血症的患儿,予降脂药物如他丁类药物非诺贝特治疗[4]。对于合并单纯蛋白尿的患儿可给予血管紧张素转换酶抑制剂如赖诺普利治疗。⑷肾脏替代治疗及肾移植 对于肾衰竭患者,需肾脏替代治疗及肾移植治疗。在表X-1关于HLTRS的报道文献中,病例1的父亲、病例3、病例4均出现了肾衰竭,接受了肾脏替代治疗,且并随后接受了肾移植,且肾移植后移植肾功能一直稳定,病例4肾移植后出现单侧特发性周围神经麻痹,经糖皮质激素治疗得以改善。
4.3 其他对症治疗对于合并肺动脉高压的患儿可给予西地那非、波生坦、依前列醇治疗[14],但疗效不确切。对于严重鞘膜积液、肠套叠等患儿需外科手术治疗。对于合并严重心包积液、胸腔积液等的患儿需要引流治疗。对于合并淋巴水肿的患儿可以予压力支持治疗[6]。
5 小 结
HLTRS为罕见的遗传性疾病,但其在国内的发病率可能被低估,本文着重从该病的发病机制、临床表现、诊断及治疗这四方面进行了综述。该病的严重程度与SOX18基因突变的位点相关,严重胎儿期即死亡或发病,有幸顺利出生的患儿,虽可以达到长期存活,但多伴有严重症状性高血压,并可进展至终末期肾病,需要肾脏替代治疗或肾移植治疗。对于临床疑似病例需进一步行肾穿刺及基因检测,尽早诊断及调整治疗,或可延缓疾病尤其是肾损害进展。该病尚无明确有效的治疗方法,SOX18显性突变蛋白抑制剂或基因疗法有待进一步探索。
