H2S在Cr(111)面上吸附与解离的第一性原理研究
2024-01-19江佐禹黄本生何子涛
江佐禹, 黄本生, 2, 何子涛, 罗 霞
(1. 西南石油大学 新能源与材料学院, 成都 610500; 2. 西南石油大学 四川省玄武岩纤维复合材料及开发应用工程技术研究中心, 成都 610500)
1 引 言
随着科技的发展,人们对能源的需要越来越大.为了满足需求,越来越多的含硫油气田被开发出来,同时也带来了原油、天然气开采过程中的管道等器材腐蚀问题[1]. H2S作为一种伴生气体广泛存在于石油和天然气开采过程中,在干燥环境中不具有腐蚀性,但是一旦溶于水会使水溶液产生酸性,这种物质不仅会引起管道的氢致开裂和硫化物应力腐蚀开裂,还会导致大多数的金属催化剂失活[2-5]. 因此研究H2S在金属表面的反应过程对防腐材料的开发具有重要意义.
为了提高材料的耐蚀性,人们往其中添加Cr、Cu和Mo等合金元素. Yang等人研究Cu的添加对Ni-Co-Cr-Mo合金耐蚀性的影响,其中Cu含量为2%时,此时形成的钝化膜最厚,耐蚀性能最好[6]. Ajit研究发现,在镍基合金中加入Cr、Mo元素,在盐酸和硫酸溶液中表现出很强的均匀耐腐蚀性,同时其合金的局部耐腐蚀性高于其他的在售合金[7]. Liu等人通过对比分析添加不同Cr含量的Ni-Cr-Mo涂层在H2S下的腐蚀性能,发现随着Cr含量的增加其耐腐蚀性能显著增加,当Cr含量为15wt.%时,其耐腐蚀性能和用量是最优的[8]. 从这些研究可以看出,Cr对材料的耐腐蚀性的提升具有很大的作用. 然而令人惊讶的是关于Cr金属表面微观机理研究却非常稀缺,现仅发表了一篇在Cr表面上的研究,Anil 等人运用了DFT模拟研究了氢同位素在体心立方Cr和Cr(100)表面上的化学吸附过程[9]. 如今,有很多文献研究了H2S在金属[10-19]及其合金[20-22]表面上的吸附解离过程. H2S在这些表面上的吸附过程通常被描述成一个跨越低能垒的简单过程. 而H2S在Cr表面上的具体吸附机理并无研究. 本文选用Cr(111)面研究H2S吸附解离过程,为后续的含铬防腐材料的开发提供更深入的了解.
2 计算方法
本文采用Material Studio软件中的CASTEP模块[23]进行计算,所有计算过程都是利用自旋无限制的平面波密度泛函进行. 采用的是广义梯度近似(GGA)下的Revised-Perdew-Burke-Ernzerh(RPBE)泛函[24]进行研究. 体计算和表面计算的布里渊区积分分别使用(4×4×4)和(4×4×1)的K点网格,截断能设置为400 eV. 为了保证收敛,能量收敛准则设置为1.0×10-5eV/atom,原子作用力收敛准则设置为0.03 eV/Å,应力偏差小于0.05 GPa,公差偏移小于0.001 Å.
在基于优化的Cr晶体结构,建立了4层的Cr(111)表面模型,采用了p(2×2)的超晶胞结构,吸附物的覆盖率为1/4ML. 其中最下面两层原子被固定住,其余层的原子完全弛豫,为了保证周期性平板之间不会发生相互作用,在平板的z方向加上了15 Å的真空层. 为了保证计算的可靠性,我们计算了Cr的晶格常数(0.286 nm)与实验值(0.288 nm)非常接近,同时H2S、HS和H2的键长和键角理论值与实验值差别如表1所示,误差值均小于2.2%.
通过线性同步变换(LST)和二次同步变换(QST)方法得到反应的最小能量路径,研究H2S在Cr(111)表面上的吸附解离过程,并且确定过渡态(TS).
吸附能公式表示如下:
Eads=Eadsorbates/slab-Eslab-Eadsorbates
(1)
其中Eslab、Eadsorbates、Eadsorbates/slab分别表示洁净的表面,在气相中的吸附物分子,和具有吸附物分子的表面的能量. 由定义可知,吸附能为负值则为放热反应.数值的绝对值越大,放热能量越多,说明吸附作用越强.
对于在Cr(111)面上计算的活化能Ea和反应能△E可以表示如下:
Ea=E(TS)-E(RE)
ΔE=E(PR)-E(RE)
(2)
其中E(RE)代表反应物在Cr(111)面上初始态的总能量,E(TS)代表过渡态在Cr(111)面上的总能量,E(PR)代表最终产物在Cr(111)面上的总能量.
3 结果与讨论
我们考虑了几种不同的吸附位置研究H2S及其解离产物在Cr(111)面上的吸附:代表Cr原子顶部的位点Top,代表两个Cr原子之间的桥接位点Bridge. 如图1所示,Cr(111)面有3层原子裸露在外面,在Cr(111)面上顶部的位点就有三种,分别为:第一层Cr原子的顶部(T1)、第二层Cr原子的顶部(T2)和第三层Cr原子的顶部(T3);同时桥接位点也有三种,分别是:第一层Cr原子与第二层Cr原子之间的桥接位点(B12)、第二层Cr原子与第三层Cr原子之间的桥接位点(B23)和第一层Cr原子与第三层Cr原子之间的桥接位点(B13). 本次研究黄色圆球代表S,白色圆球代表H,其他颜色原子均为Cr,仅考虑了垂直吸附.
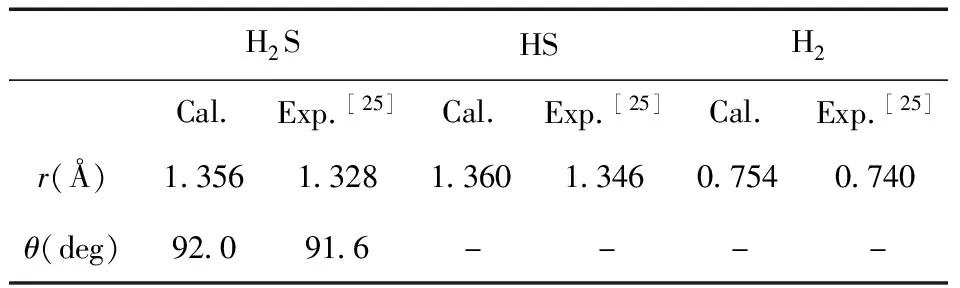
表 1 气相 H2S、HS、H2 的几何参数
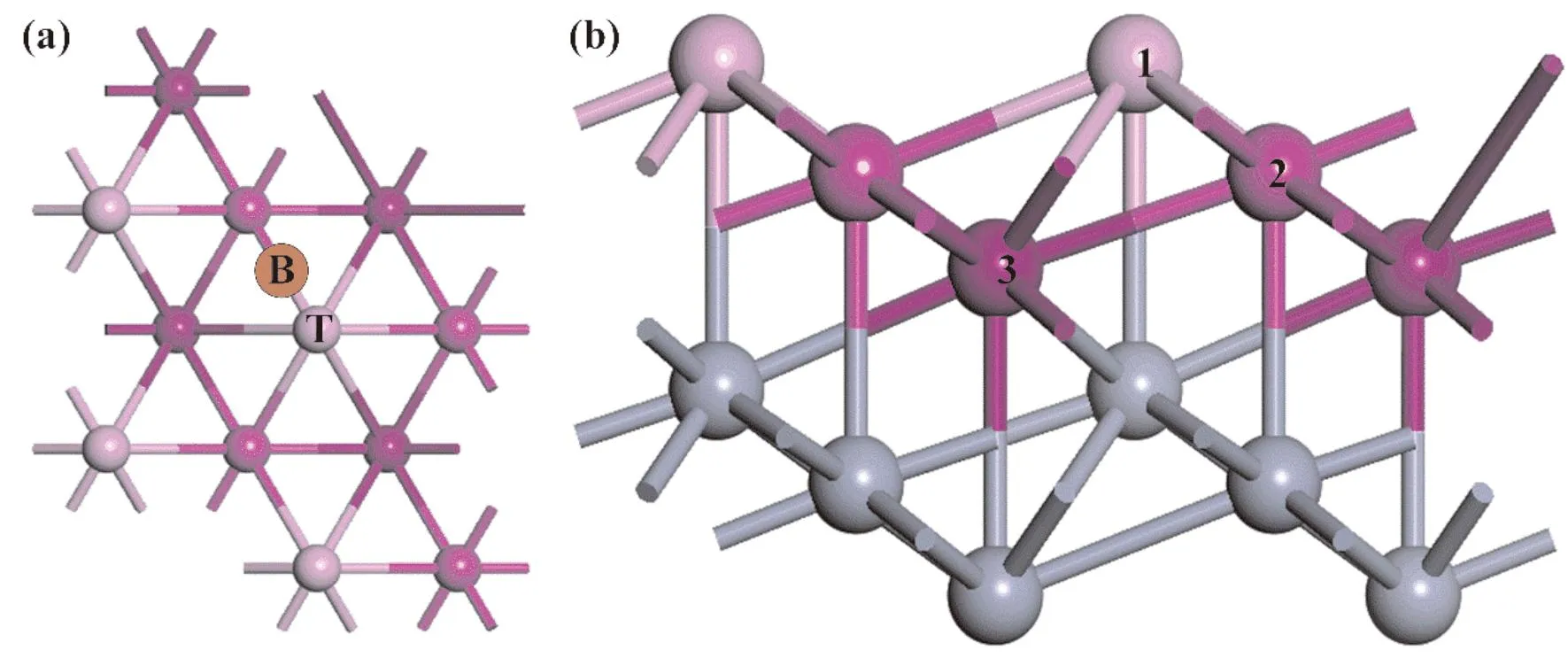
图 1 (2×2)Cr(111)表面示意图:(a)俯视图,(b)侧视图. T 代表顶部站点,B 代表桥接站点. 侧视图中的1、2和3表示Cr(111)表面上的第一层、第二层和第三层原子.Fig. 1 (2×2)Cr(111)surface schematic diagram:(a)top view,(b)side view. The T stands for the top site and the B for the bridge site. 1,2 and 3 on the side view represent the first,second and third layer atoms on the Cr(111)surface.
3.1 H2S及其解离产物的单一吸附情况
对于H2S及其解离产物在Cr(111)表面单一吸附的结构图如图2所示,相应最优吸附位置、吸附能以及结构信息如表2所示.

表2 H2S及其解离产物在Cr(111)面上的最优吸附结构参数以及吸附能
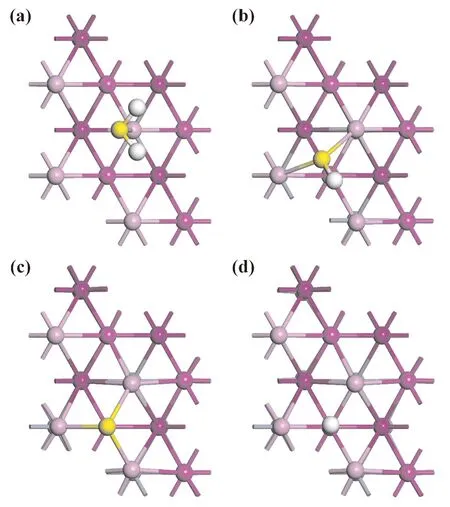
图2 H2S及其解离产物在Cr(111)面上的最优吸附位:(a)H2S,(b)HS,(c)S,(d)HFig. 2 The optimal adsorption sites of H2S and its dissociation products on the Cr(111)surface:(a)H2S,(b)HS,(c)S,(d)H
研究发现,H2S的稳定吸附位点是T1位点(图2(a)),而吸附在其他位点是都发生了不同程度的分解和偏移. 当H2S吸附在T1位点,由一开始的垂直吸附变成了倾斜吸附,这意味着顶部的倾斜吸附更加稳定. 同时H-S键和∠H-S-H发生了不同程度的增大,表明Cr表面将H2S活化,导致H-S键被激活且强度减弱.
对于HS,由于其的不稳定性,并没有在实验上观察到其吸附在金属表面上的吸附结构. 根据吸附能来看,在T1位点的吸附能最小为-2.81eV,在B23位点的吸附能最大为-3.84 eV. 并且HS吸附在T2、T3和B12位点时,都往Cr(111)表面上的B23位点发生了偏移. 而当HS吸附在B13时,往Cr(111)表面上的T3位点发生偏移. 这进一步说明了B23位点是HS在Cr(111)表面上最稳定的吸附位(图2(b)). 根据H-S键吸附前后对比,结果表明,H-S键更倾向于倾斜吸附于基体表面,H-S键长由大气中1.353 Å延长到了1.373 Å,说明Cr表面活化了H-S键导致H-S键增长,H-S键减弱且容易断裂.
对于S和H,S在Cr(111)面上的6种吸附位置都较为稳定,均呈垂直吸附于基体表面上,但当S处于Cr(111)面B12位点和B13位点时,经过结构优化后分别转移到T2位点和T3位点;其中在T2位点的吸附能是最大的(图2(c)),为-6.76 eV. 吸附时与周围Cr原子形成了4个S-Cr键,键长分别为2.343 Å,2.496 Å,2.496 Å和2.499 Å. 而H在Cr表面的最优吸附位为T2位点(图2(d)),仅与一个Cr原子成键,吸附能和键长分别为-0.45 eV和1.761 Å.
为了进一步理解H2S及其解离产物在Cr(111)面上的单一吸附情况,我们绘制了H2S、HS、S和H在Cr(111)面吸附前后的偏态密度图(PDOS)如图3所示. 从态密度图中可以看出,吸附前后S元素的s轨道波峰、p轨道波峰和H元素的s轨道波峰均显著往低能级方向移动,说明H2S及其解离产物在Cr(111)表面上均为化学吸附.

图3 H2S及其解离产物吸附前后的偏态密度图:(a)H2S,(b)HS,(c)S,(d)HFig. 3 Partial density of states (PDOS)plots of H2S and its dissociation products before and after adsorption:(a)H2S,(b)HS,(c)S,(d)H
根据图3(a)、图3(b)和图3(c)中吸附后的态密度图可以看出部分S-p轨道电子与Cr-d轨道电子处在同一能级. 表明H2S、HS和S吸附时S-Cr键的形成主要是S-p轨道电子和Cr-d轨道电子的杂化共轭作用. 由图3(d)吸附后的态密度图可以看出少量的H-s轨道电子与Cr-d轨道处于同一能级,H吸附时H-Cr键的形成主要是H-s轨道电子和Cr-d轨道电子的杂化共轭作用.
通过对比S、HS和H2S吸附时态密度图中S-p轨道与Cr-d轨道的重叠面积可以发现,说明S吸附体系上S-p轨道与Cr-d轨道的杂化共轭作用是最强的,其次是HS分子,最后是H2S分子. 进一步证明了在Cr(111)表面上的吸附强度大小排序:S>HS>H2S.
3.2 HS/H和S/H在Cr(111)面上的共吸附情况
图4(a)显示了HS和H在Cr表面的共吸附构型,HS处于B23位置,H处于T2位置. 与HS/H单一吸附的情况不同的是,在共吸附情况下,S-Ni键长和H-Ni键长相对于单一吸附情况下均有所增加,而H-S键长从1.373 Å降到了1.371 Å略有降低. 其共吸附的吸附能为-4.284 eV,小于单一吸附情况下HS与H吸附的吸附能总和-4.309 eV. 说明H与HS之间在Cr(111)面上吸附时具有一定的相互作用.
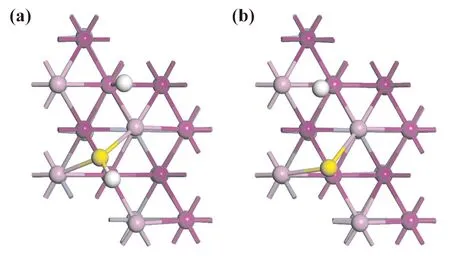
图4 Cr(111)面上的共吸附构型:(a)HS/H,(b)S/HFig. 4 Co-adsorption configurations on Cr(111)surface:(a)HS/H,(b)S/H
图4(b)显示了S和H在Cr表面的共吸附构型. S处于T2位置,H处于S相邻的T2位置,不同于单一吸附情况的是,S只与周围3个Cr原子成键,S-Cr键长(2.250 Å-2.552 Å)不同于单一吸附情况的S-Cr键长(2.343 Å-2.499 Å). H在T2位置也是倾斜吸附状态,且H-Cr键的键长从1.761 Å增加到了1.767 Å. 在Cr(111)表面,S与H的吸附能为之和为-7.205 eV,与共吸附能(-6.931 eV)不同. 这种现象说明S与H之间在Cr(111)表面上也具有一定的相互作用.
4 H2S的解离步骤
我们在确定了H2S及其解离产物在Cr表面上的稳定吸附位置之后,通过LST/QST方法确认了H2S在Cr(111)面上解离过程的最小能量路径如图5所示,同时绘制了H2S在Cr(111)面上解离的能量分布图(图6). H2S分子的解离步骤有两步,分别为:H2S→HS+H,HS→S+H两个步骤. 由图4可知,H2S首先倾斜吸附于第一层Cr原子的顶位(T1),S与一个Cr原子成键,随后发生了第一步解离. 第一个H从H2S上脱离,向二层Cr原子的顶部位点(T2)方向移动,此时H还没有与Cr原子成键. 同时HS也向B23位置偏移. 在TS1之后,H与一个Cr原子成键,呈倾斜吸附. 而HS也完全倾斜吸附在B23位点上,其中S与周围三个Cr原子成键. 在第二步解离过程开始时,HS的最优稳定位置是处于第二、三层Cr原子之间的桥接位点(B23),随着解离的发生,H往另外一个二层Cr原子的顶位方向(T2)偏移,形成TS2,这时H-S键还没发生断裂. 随着反应的继续进行,H-S键发生断裂,H移动到了二层Cr原子的顶部位点,与一个Cr原子成键,呈倾斜吸附;S移动到了另一个二层Cr原子的顶部位点,与周围的3个Cr原子成键. 在第一步解离过程中,反应的活化能为1.65 eV,反应为放热反应(-1.95 eV),说明第一步解离反应在热力学与动力学层面上是可以自发进行的. 第二步解离反应的活化能较第一步反应的活化能偏低,为0.82 eV,反应能为-0.95 eV,说明第二步解离反应比第一步反应更加容易进行.

图5 H2S的解离路径Fig. 5 The dissociation pathway of H2S
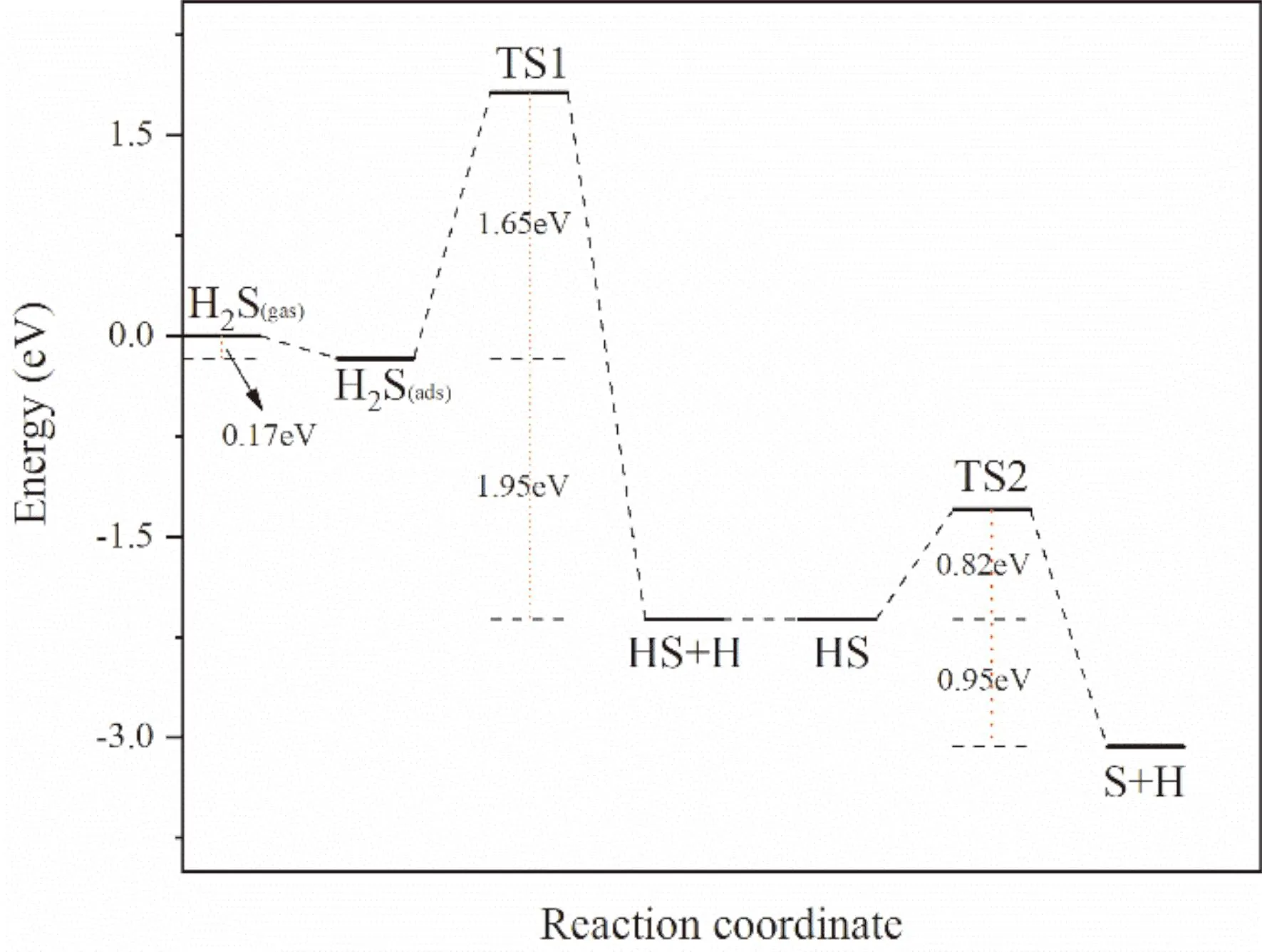
图6 H2S在Cr(111)面上解离的能量分布图Fig. 6 Energy distribution of dissociation of H2S on Cr(111)surface
5 结 论
(1)对于单一吸附在Cr(111)面的情况,H2S偏稳定吸附在第一层Cr原子的顶部位置,HS偏稳定吸附在B23位置,而S与H都偏稳定吸附在T2位置,这几种物质在Cr(111)面上的吸附能大小顺序为:S>HS>H>H2S.
(2)H2S、HS、S和H能稳定吸附在Cr(111)面主要是因为S-p轨道、H-s轨道和Cr-d轨道发生杂化共轭作用. 同时通过S-p轨道与Cr-d轨道的重叠面积进一步证明S的吸附能最大,HS其次,H2S最后.
(3)在共吸附情况下,HS的吸附情况变化不大,两个共吸附构型中H都变成了倾斜吸附. S吸附时与单一吸附情况下不同,倾斜吸附在T2位置,仅与周围3个Cr原子成键. 两种共吸附情况的吸附能均与单一吸附情况有所不同,说明共吸附时吸附物质之间存在相互作用.
(4)H2S在Cr(111)面上的解离过程是放热的,反应能为-2.90 eV. 同时第一步解离的活化能为1.65 eV,约为第二步解离活化能(0.82 eV)的两倍. 说明H2S若想在Cr表面上发生解离过程,首先需要跨越一个相对较大的能垒,才能进一步解离.
