单核苷酸多态性(SNP)分析在细菌分子分型中的应用
2024-01-15杨富萍杨向东于彬彬高子厚
杨富萍,杨向东,于彬彬,高子厚
细菌分型是指对分属同一种或亚种的细菌菌株采用特定的试验方法进行基因特征分析,并根据菌株的流行病学数据,揭示被研究菌株之间的基因联系[1]。细菌分型及检测技术包括表型分型技术、基因分型技术、色谱与质谱技术、细菌自动化鉴定技术,但常规的分型技术主要是指表型分型技术,有生物化学分型、细菌素分型、耐药普分型等,但此类技术往往具有重复性低、操作复杂,以及分辨力较低等弊端,存在较大的局限性。近年来,分子生物学技术的快速发展,为细菌分型提供了更有力的手段,将细菌分型提高到分子层次。目前对细菌分子分型技术包括脉冲场凝胶电泳技术(PFGE)、多位点可变数目串联重复序列分析(MLVA)、多位点序列分型(MLST)、聚合酶链式反应(PCR)、限制性片段长度多态性分析技术(RFLP)、实时定量PCR(real-time PCR)[2]等。一种好的分型技术须具有操作简单、稳定性好、成本低、分辨率高、实验结果易于理解、便于宣传等优点。在众多微生物分子分型方法中,SNP技术作为一个新型的分型技术,在微生物分子流行病学方面得到逐步肯定与发展。本文介绍了该技术在细菌分子分型中的应用。
1 SNP技术研究概况
SNP是指基因组DNA序列中由单个核苷酸(A,T,C和G)的突变而引起的多态性,从理论上来看每一个SNP 位点都可以有4 种不同的变异形式:转换、颠换、插入或缺失,且发生变异频率大于1%[3],但通常发生的变异只有两种,即转换和颠换,比例为2∶1[4]。出现在编码区时,可分为同义SNP(sSNP)和非同义SNP(nSNP),sSNP不能修改其编码的氨基酸,其多态性客观的反映了细菌的进化状况,成为研究致病性细菌群体遗传学、微进化和系统发育学的金标准[5]。而nSNP会导致氨基酸的内部结构发生变化,进而改变蛋白质的相关功能,特别是出现在结构功能区域中的SNP;出现在非编码区时,尽管此区SNP的数量众多,但不会使个体的表型性状发生改变,却可以成为群体遗传与个体进化研究中的重要遗传标志。SNP最初在人类基因组中被发现,占据人类基因组多态性的90%[6]。90年代初,SNP首次于1994年在人类分子遗传杂志上出现,之后于1996年由Lander ES第一次提出SNP技术作为第3代遗传标记[7],在人类、作物、动物等多个物种中研究逐步加深[8]。目前通过许多检测方法,如单链构象多态性(SSCP)、限制性酶切片段长度多态性、DNA测序、基因芯片法等已经能获得个体的SNP[9],SNP分析能提供完整的数据信息和具有极高的分辨率,目前不仅在个体诊断治疗、临床医学、群体遗传学、法医学鉴定等研究领域中发挥巨大作用外,而且在鼠疫耶尔森氏菌、布鲁氏菌、炭疽芽孢杆菌、结核分枝杆菌等致病菌的研究领域也得到广泛应用。
2 SNP技术的优缺点
目前对疾病暴发的分子溯源和流行病学调查,传统的表型分型方法所提供的遗传学信息有限,且耗时费力,已远不能满足实际需求,而基于核酸序列的各种分子分型方法迅速发展弥补了传统方法的不足。SNP技术与其它分子分型技术在细菌分型中的应用情况见表1。

表1 几种常用细菌分子分型方法对比
与其他分子分型方法相比,SNP技术具有以下优点:1)方便可行的实验设计:开发合适的分析软件,持续改进数据库,能在短时间内完成实验设计;2)数量众多,分布广泛。在人类基因组中,平均每一千个碱基就会出现一个SNP;3)遗传稳定,突变率仅为10-9;4)适于大规模、快速检测。基因组片段的长度对分析没有影响,操作简便,利于自动化,缩短了研究时间;5)等位基因的大小易于预测,便于基因分型;容易受到选择条件、环境等因子的干扰,特别适用于研究近亲的微观进化特性[10]。总之,SNP技术简便、自动化程度高,通量大,快速,分辨率极高,是其他分子分型技术所无法比拟的。
式中:当时,Fij表示Rij相对于产生的损失,Rij越大,产生的损失越大;当时,Fij表示Rij相对于产生的损失,
3.2 鼠疫耶尔森菌 鼠疫耶尔森菌所致的鼠疫是自然疫源性烈性传染病,我国传染病防治法将其列为甲类传染病,在二战时期霸权主义曾把鼠疫菌作为重要的生物战剂之一。2010年Morelli等[17]对来自全球的286 株鼠疫菌进行了比较基因组学分析,筛选出分离株集合中的17个完整基因组序列和933个SNP序列,利用这些序列推测了鼠疫菌的历史传播途径——鼠疫菌在中国或中国附近进化,并通过多次辐射传播到欧洲、南美、非洲和东南亚,目前来自美国的分离株都显示了一种辐射,来自马达加斯加的分离株代表了第2种辐射。2013年Cui等[18]对分离自亚洲、欧洲、美洲和非洲等地的133株鼠疫菌菌株进行了全基因组研究,在这些鼠疫菌的核心基因组中共找到2 326个SNP位点,该 SNP集定义了鼠疫菌自其最近的共同祖先以来的谱系,利用这些数据构建系统发育树,推测了鼠疫菌的传播路线——Angola菌株祖先节点的分化时间与第一次鼠疫世界大流行的时间吻合,目前世界鼠疫菌四大种系分支的形成与鼠疫第二次世界大流行(黑死病)密切相关;鼠疫菌种群结构及菌株的地理分布规律表明,古代商路(丝绸之路、唐蕃古道和茶马古道)在鼠疫的传播中发挥了重要作用,而青藏高原东部可能是鼠疫菌最早的起源地之一。2019年Li等[19]采用全基因组SNP分析对2018-2019年从内蒙古分离的40株鼠疫菌进行系统发育研究,发现内蒙古不同地区的鼠疫菌株不是由同一种动物传播引起的,而是通过不同的动物传播引起的。通过以上研究表明,SNP技术将有助于进一步探索鼠疫菌的进化关系史及其历史传播模式。
3.4 炭疽芽孢杆菌 炭疽芽孢杆菌是一种危害极其严重的“生物武器”,SNP是检测和区分炭疽杆菌的重要诊断标志物。2007年Van等[25]对88个分离自全球的炭疽杆菌菌株及1株Ames菌株(基因型62)进行SNP测定分析,共鉴定出3 500个SNP,发现其中6个 SNP对 Ames 菌株具有高度特异性,能将Ames菌株与 88 个独特的炭疽杆菌菌株区分开来。2021年Pisarenko等[26]将1956-2018年在东西伯利亚和远东地区分离的15株炭疽杆菌的基因组序列与GenBank上公布的219个炭疽杆菌基因组序列进行比较分析,共检测到6 400个SNP,利用检测到的SNP构建系统发育树,将所研究的菌株分为5个不同的遗传类群。2022年Anisimova等[27]对从土壤样品和动物尸体中获得的 7 株炭疽杆菌进行SNP分析,将所研究的菌株分成四个簇。最大的集群由鞑靼斯坦共和国(6、7号菌株)和乌里扬诺夫斯克(5号)分离的三个菌株形成。第二大集群为从塔吉克斯坦收集的2号和4号菌株。剩下的由在车臣-印古什ASSR和在库尔干地区发现的1号和3号菌株形成。随着SNP技术的改进,由于SNP技术的进步,对炭疽杆菌的分辨力和再现性也在逐步增强,目前已经是标准的炭疽杆菌分型技术,为其鉴定溯源提供了有力工具。
3 SNP技术在细菌分型中的应用
3.3 结核分枝杆菌 结核分枝杆菌是引起结核病的病原菌,以肺结核最为多见,至今仍为重要的传染病,居各种疾病死亡原因之首,据WHO报道,每年至少有300万人死于该病。2002年Gutacker等[20]对4株已完成全基因组测序的结核分枝杆菌进行比较分析,得到了230个nSNP,并利用这些SNP对来自全球的432株结核分枝杆菌进行了基因分型,最终将这些菌株分成8个基因型群。2006年Filliol等[21]使用 212 个SNP标记分析了全球323株结核分枝杆菌集合,发现SNP多样性很高,96% 的 SNP 基因座处于完全连锁不平衡状态,通过聚类分析将这些菌株分成5个亚组。北京菌株是全球结核分枝杆菌最成功的基因型之一,根据现有的基因分型方法,发现其具有高度同质性。Schürch等[22]、Luo Tao等[23]分别于2011、2012年先后对北京菌株进行SNP分析,发现北京毒株多次在全球传播,由北京基因型引起的结核病流行至少部分是由现代移民模式驱动的。非洲结核分枝杆菌(Maf)共有L5和L6两个谱系,它们是结核分枝杆菌复合体(MTBC)的成员。2022年Balamurugan等[24]利用全基因组SNP的方法估计了380个Maf样本的遗传多样性,观察到基于谱系的聚类(L5和L6)具有不同的亚聚类(L5.1.1和L5.1.2,L5.2.1-L5.2.4,L6.1-L6.3),同时发现携带多个(亚)谱系特异性“核心簇”SNP的基因,如Lys117Asn、Val447Met和Ala455Val,分别存在于L6、L6.1和L5中,暗示这些SNP与选择性优势或宿主适应有关。以上研究表明SNP分析已广泛应用于结核分枝杆菌的基因分型,将有助于未来对该菌的分子流行病学和系统发育研究。
采用SPSS 21.0软件进行分析,组间比较采用有序分类变量两组独立样本的秩和检验。其中P<0.05认为差异具有统计学意义。
年终的回馈客户活动,部门领到厂家的30块Swatch赠表。我在晨会上建议,因为赠品有限,最好是配合销售,手表赠送给购买服务器或批量PC机客户,末了,我自鸣得意地说:“Swatch是货真价实的瑞士名表,送给大客户也算是好钢用在了刀刃上。”
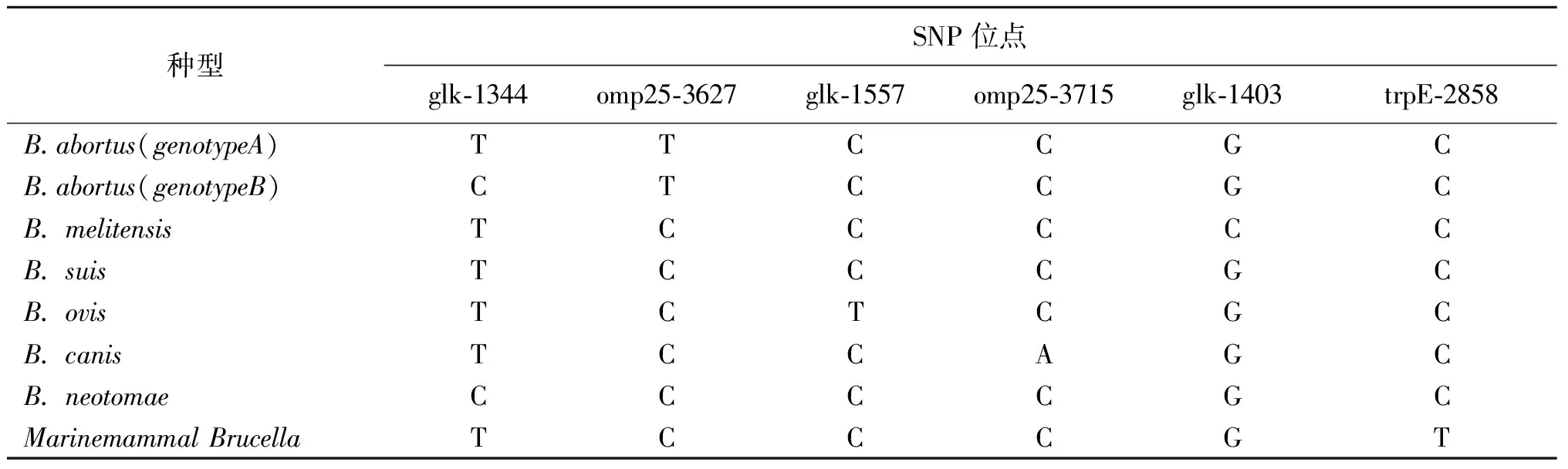
表2 区分6种布鲁氏菌和海洋哺乳动物布鲁氏菌菌株的特定SNP位点
通常可以用辛普森生物多样性指数(Simpson’s diversity index,SDI)来衡量SNP的分辨率。2018年Janowicz Anna等[11]对布鲁氏菌的研究显示,108株布鲁氏菌被分为51个MLVA-16型、55个cgMLST型和60个SNP型,分型的鉴别能力比较表明,基于SNP的方法优于其他两种方法,SDI为0.922,95%置信区间(CI)为0.866~0.978。cgMLST的SDI计算为0.815(95%CI,0.685~0.945),MVLA-16的SDI为0.674(95%CI,0.505~0.843)。2020年Ksibi等[12]对175株肠炎沙门氏菌进行几种分型方法比较,同样得出基于SNP的方法优于其他方法,SDI为0.997,MLVA为 0.889,cgMLST为0.785,PFGE为0.518。
3.1 布鲁氏菌 布鲁氏菌是导致布病的人兽共患病原菌,主要是通过接触或者饮食等方式感染传播给人类。2007年Scott等[13]利用SNP分析对330株布鲁氏菌菌株的glk、omp25和trpE基因片段进行测序分析,通过序列比对共筛选出6个特异性的SNP位点(表2),能够在种水平上鉴定布鲁氏菌,并最终在超过500 株菌株中得到验证。2014年谭鹏飞等[14]为区分中国B.bovis疫苗株A19与野生株,采用基因组测序的方法,共筛选出A19基因组29个SNP位点,选取ClpX G825-C825、LysR A605-C605、Omp2b G503-A503 3个SNP位点,与布鲁氏菌常见种、标准参考菌株基因组SNP进行比较,发现这3个SNP位点仅存在于A19(S19)基因组上,可用这3个SNP位点鉴别我国布鲁氏菌疫苗株A19与野生菌株。2015年Tan等[15]对来自马来西亚和菲律宾的布鲁氏菌分离株进行SNP分析,将分离株分为5个基因型:地中海菌株,被确定为基因型 I,所有亚洲菌株与SEA菌株聚集成基因型 II,基因型 III 代表非洲血统,基因型 IV 代表欧洲血统,基因型 V 代表美洲血统。2021年Zhao等[16]对2名男性血培养物中分离的布鲁氏菌进行SNP分析,发现所研究的2株菌株属于同一基因型,但属于不同的亚群,1株与从近东(塞浦路斯)分离的菌株最相似,1株与从中亚(俄罗斯:图瓦共和国)分离的菌株最相似,表明了2株菌株各自的起源。通过以上研究发现SNP分析是布鲁氏菌属种内鉴别的有力工具,可为追溯分析提供有关地理来源的有用信息,能对疫苗株和野生株进行鉴别。
SNP技术自问世以来发展迅速,因其自身的特性,在细菌分型与进化、流行病学调查研究中得到广泛应用。
尽管SNP技术在细菌的分型上应用很普遍,但它本身也具有某些缺陷:1)SNP研究要求操作人员具有较高的专业水平,分析数据时需要强大的生信分析能力;2)对研究菌株的WGS数据品质要求非常高,为了保证基因组数据的准确度和一致性,需要选择很严谨的技术标准,如SNP之间的最小覆盖范围和允许间隔。
为打赢脱贫攻坚这场硬仗,积极引进培育农业经营主体,大力发展农业产业,石柱县探索并构建了“1+4”资产收益扶贫政策体系,即出台了《关于推广资产收益模式促进经营主体与贫困户利益联结共同发展的实施意见》1个实施意见,并配套股权收益扶贫、基金收益扶贫、信贷收益扶贫和旅游收益扶贫4个实施方案,多方整合财政补助资金,最大限度提高涉农资金的使用效益,带动贫困户长期稳定增收。
3.5 金黄色葡萄球菌 金黄色葡萄球菌是常见的食源性致病菌,广泛存在于自然环境中。在适当的条件下,能够产生肠毒素,引起食物中毒。2006年Huygens等[28]对在澳大利亚昆士兰州东南部收集的 391 个金黄色葡萄球菌分离株进行了SNP基因分型。共使用了8个SNP集,提供了整个金黄色葡萄球菌SNP数据库的辛普森多样性指数(SDI)为0.95,并定义了61个现存的金黄色葡萄球菌基因型和主要克隆复合物。2014年Holmes等[29]利用SNP分型方法对代表苏格兰流行的EMRSA-15基因型104株金黄色葡萄球菌分离株进行了系统发育分析。在这些菌株的核心基因组中共鉴定出904个高质量SNP,研究选择了对EMRSA-15亚型具有判别意义的17个SNP进行分析。2020年Pirolo等[30]对67株耐甲氧西林金黄色葡萄球菌序列398(LA-MRSA ST398)菌株进行了全基因组测序分析,发现LA-MRSA ST398在意大利南部的引入和传播存在两种并发模式,一种是通过与包括丹麦和法国在内的其他欧盟国家进行仔猪交易,多次引入LA-MRSA ST398菌株;另一种是独立的ST398克隆体的扩展,特别是在与其他意大利农场交易动物的农场。通过分析证实SNP是一种快速、稳健、可重复(分析内变异系数<25%)的鉴别技术,可以潜在地应用于金葡菌鉴定及溯源。
3.6 在其他细菌分型中的应用 随着SNP技术在生物分子分型上的应用,也有研究人员将SNP技术应用于其他细菌研究中。例如,利用WGS数据与流行病学数据一起分析得出2017-2018年南非李斯特菌病暴发的源头是Enterprise Foods生产的即食加工肉制品[31],使用SNP分型方法对全球伤寒沙门氏菌血清型进行分型,有助于全球流行病学分析[32],采用宏基因组学联合全基因组SNP分析等其他检测技术对志贺菌暴发进行病原学分析[33],使用SNP技术对大肠埃希氏菌进行地理溯源研究[34],通过SNP基因分型推测麻风分枝杆菌传入我国可能的2条路线[35]。
随着设备运行时间的增加,使用中逐步出现一些问题。选煤厂对这些常见问题进行了分析总结,掌握了故障的处理方法,同时根据现场实际进行了改造,使设备运行得以更加完善。
4 SNP生物信息学分析数据库及在线工具的应用
随着高通量测序技术的飞速发展以及相应成本的下降,SNP数据信息日渐丰富,由此形成了许多关于致病菌的SNP信息库。2010年McKenna A等[36]开发了基因组分析工具包(GATK),这是一个结构化的编程框架,旨在利用MapReduce的功能编程理念为下一代DNA测序仪开发高效、稳健的分析工具。GATK编程框架使开发人员和分析人员能够快速和轻松地编写高效和健壮的NGS工具,其中许多已经被纳入大规模测序项目,如1000基因组项目和癌症基因组图谱。2011年Winsor GL等[37]为了提供一个高质量、带注释的基因组资源,便于假单胞菌物种进行跨菌株和跨物种基因组比较,开发了假单胞菌属基因组数据库,有助于分析密切相关菌株之间的表型变异,以及更广泛的群体基因组学和进化研究。2013年Chattopadhyay S等[38]为了分析细菌菌株之间的遗传差异(基因存在/缺失和核苷酸多态性),了解细菌发病的分子机制和选择新的抗菌治疗的靶点,建立了微生物变异组数据库,包括22株大肠杆菌和17株肠炎沙门氏菌的完全测序基因组的核心蛋白编码基因的点突变数据。这些资源数据库的建立极大地方便了人类病原体的研究,并推动SNP技术的发展。
5 展 望
一种好的分型方法检测原理要清晰严谨,通量大,具有一定的自动化规模,技术上简单快捷、成本低、精度高、可靠性强、分型性能好、结果易于解释等特点。比较而言,SNP技术是相对较为理想的一种方法,随着相关技术的不断创新,方法的改进及各类编程语言和专门化软件的应用使得数据分析更为便利,SNP分型技术也将向更加高效、准确、经济的方向发展。该技术具有操作简单、快捷、点位量大、范围广、通量高、分辨率高等优点,被广泛应用于病原体分型。现阶段第二代和第三代测序技术均得到了广泛应用,基于细菌全基因组序列的SNP研究越来越多,大量细菌SNP数据库也在陆续建立和完善,有利于全世界范围内的菌株进行参比,进一步深化人类对各种致病菌遗传进化、溯源、分子流行病学等方面的认识,有效提升人类防控疾病的水平。SNP技术作为新一代分子标记技术具有不可替代的优势,在许多致病菌的系统发育研究中发挥着不可取代的作用。
利益冲突:无
