基于整合生物信息学分析识别骨肉瘤干性相关基因
2024-01-11王思乔缐述源黄润之
符 晴,王思乔,缐述源,黄润之,张 杰
(1.同济大学医学院,上海 200092; 2.上海长海医院烧伤外科,上海 200433)

具有高度自我更新和分化能力的癌症干细胞(cancer stem cells,CSCs)被认为是癌症侵袭和转移的根源,但其潜在机制尚未被完全阐明[6]。此外,患者治疗反应和生存时间的差异提示了OS肿瘤细胞的异质性。恶性肿瘤的异质性是肿瘤诊治的关键和难点,不同基因型及表型的恶性肿瘤细胞表现出不同的生物学行为。最新的研究发现OS中癌症干细胞与患者耐药性和肿瘤发生有关[7]。基于mRNA测定的干细胞指数(mRNA expression-based stemness index,mRNAsi)是衡量肿瘤细胞与干细胞之间相似性的指标,可以被用来量化肿瘤干细胞,亦称为肿瘤干性指数。大量研究表明,mRNAsi在多种肿瘤中被作为评价患者生存预后和肿瘤分型的有效指标,如结直肠癌、乳腺癌、头颈部鳞状细胞癌等[8-10]。然而目前仍然没有研究基于mRNAsi来分析OS的肿瘤干性。近年来,单细胞测序(single cell RNA sequencing,scRNA-seq)技术的发展为肿瘤细胞异质性的分析提供了新方向[11]。单细胞测序可从基因组学、转录组学、表观遗传学以及多组学对肿瘤微环境进行分析,能进一步揭示肿瘤内部的细胞异质性,研究肿瘤的发生、发展及转移过程,阐明肿瘤患者耐药机制,绘制肿瘤微环境的单细胞图谱[12-13]。本研究采用整合生物信息学分析的方法,识别转移性骨肉瘤中干性相关基因(stemness-related genes,SRGs),在单细胞水平深入地探究OS肿瘤细胞的异质性,为实现转移性OS分子治疗的转化医学研究提供理论依据。
1 材料与方法
1.1 数据来源及其预处理
OS患者的RNA测序(RNA sequencing,RNA-seq)数据和临床信息(年龄、性别、种族、存活时间和转移状态)来源于TARGET数据库(https:∥ocg.cancer.gov/programs/TARGET)。在GEO数据平台上(https:∥www.ncbi.nlm.nih.gov/gds)下载OS单细胞测序数据文件,GPL24676(平台注释文件)与矩阵文件GSE152048(包含11个OS患者样本)。删除临床信息不完整以及不符合纳入、排除标准的OS患者数据,最终筛选出87例OS患者,其中65例为原发性OS患者,其余22例为转移性OS患者。数据截至2020年3月。为了减少测序误差和批次效应,用微阵列分析线性模型软件包来校正已筛选的RNA-seq数据[14]。在R3.5.1环境下,将OS样本的转录组测序结果合并为1个矩阵,使用“gen-code.v22.annotation.gene.probeMap”文件注释转录组测序数据,将基因名从Ensemble ID转换为基因符号,“edgeR”包对转录组测序的Counts数据进行标准化。
1.2 获取基于mRNA表达的干性指数
干性指数mRNAsi与肿瘤去分化程度(肿瘤干性)密切相关。并且mRNAsi值越高,患者肿瘤干细胞活性越高,发生转移的可能性也越大。为了估计OS患者的肿瘤干性,基于OS患者的mRNA表达水平,使用一类逻辑回归(one-class logistic regre-ssion,OCLR)机器学习算法计算上述87个OS患者的mRNAsi[15]。
1.3 干性指数关联性分析
皮尔逊关联性分析被用于筛选出与mRNAsi明显线性相关的基因[16]。mRNAsi(记为X)与基因(记为Y)之间的皮尔逊相关系数r定义为二者之间的协方差和标准差的商。二者协方差的计算公式如下:
其中E(X)和E(Y)分别代表二者的期望,也就是平均值。方差的计算公式如下:

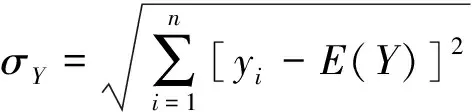
最后通过计算相关系数r来分析二者之间的关联性及关联强度。计算公式如下:
相关系数r>0.5并且具有统计学意义(P<0.05)的基因被定义为干性相关基因(Cor-sig gene),其高表达水平可能与肿瘤干细胞的自我更新能力与恶性生物学行为密切相关。
1.4 Cox回归分析
Cox回归分析是一种半参数分析方法,用于研究各种因素对患者生存期长短的影响。本研究使用R软件包中的“Survival”功能,基于OS患者的表达谱,应用单变量Cox回归分析估计每个基因的表达水平高低对患者生存时间长短的影响。风险函数用h(t)表示,计算公式如下:
表示在t时刻OS患者死亡的瞬时发生率。随后进一步构建Cox比例风险模型,计算公式如下:
lnh(t)=lnh0(t)+β1X1+…+βiXpi+…+βpXp
其中Xi是协变量,反映基因i的表达水平,βi是回归系数,h0是基准风险,即当所有Xi=0时的事件风险。随后分析特定基因的风险比(hazard ratio,HR),计算公式如下:
HR=eβi
如果值βi>0,即风险比>1,表示随着基因表达水平的增加,患者死亡的风险也逐渐增加,于是生存期减少。HR>1表示该基因的表达水平高是危险因素,可能会减少患者的生存时间。单变量Cox回归分析中HR>1并且生存时间差异具有统计学意义(P<0.05)的基因被进一步纳入多变量Cox回归分析。分析的变量包括HR,性别、年龄、原发部位、转移状态。多变量Cox回归分析中预后价值显著(P<0.05)的基因被定义为Cox风险相关基因(Cox-sig gene)。
1.5 Kaplan-Meier(KM)生存分析
KM生存分析方法即乘积极限法,由Kaplan和Meier于1958年提出,因此被命名为Kaplan-Meier法或简称为KM法[17]。作为当前生存分析最常用的非参数方法,KM法根据临床数据对患者的生存时间进行分析和推断,研究生存时间和终点事件(如死亡事件)与诸多影响因素之间的关联程度。临床数据有两种类型。(1) 完全数据:所有观察对象的生存时间或发生终点事件的具体时间都被完整记录;(2) 删失数据:观察对象在研究结束前发生了研究之外的其他事件或生存结局(失访、退出、终止等),无法明确记录到发生终点事件的生存时间。
为研究基因的表达水平与OS患者的生存相关性,根据患者特定基因表达水平将OS患者分为高表达组和低表达组,在每个OS患者发生死亡事件的时间点上,进行生存概率的计算(观察终点:12年)。其中生存概率指某时间段(tk)开始时存活的OS患者至该时间段结束时仍然存活的可能性大小,以p(tk)表示,计算公式如下:
p(tk)=tk结束时OS患者存活例数/
同组tk初始OS患者例数
由于存在删失数据,本研究假定删失事件在观察时间内各个时间点等机会发生,分母改用校正观察例数,计算公式如下:
校正观察例数=观察初始OS患者例数-观察期内删失OS患者例数/2
随后计算生存率(survival rate),以S(tk)表示,指OS患者于tk结束后仍存活的概率,计算公式如下:
S(tk)=P(T>tk)=p(1)×p(2)×…×p(tk)
本研究的KM生存曲线以生存时间为X轴,以生存率为Y轴,使用R软件包中的“Survival”功能绘制。KM生存曲线展示了生存率随时间变化的趋势,其斜率越高表示患者生存率下降越快。最后以对数秩检验法分析两组曲线之间的差异。两组总体生存曲线差异有统计学意义(P<0.05)的基因被定义为KM生存相关基因(KM-sig gene)。
1.6 绘制韦恩图(Venn diagram)
基于上述分析结果,Cor-sig gene、Cox-sig gene、KM-sig gene三者交集内的基因被定义为关键基因。在线绘图工具jvenn被用以对结果进行可视化(http:∥www.bioinformatics.com.cn/)[18]。
1.7 scRNA-seq数据标准化
将scRNA-seq数据进行前期的整理和校正,对重复基因的表达水平取平均值。采用“Seurat”R软件包中的“NormalizeData”算法对基因表达矩阵进行标准化。使用“harmony”R软件包去除数据的批次效应。使用“Percentage Featureset”函数来计算线粒体基因的百分比,并以此为标准来过滤数据(线粒体基因的百分比<5%,线粒体基因数量>50)。提取细胞间变异系数较大的基因进行后续分析。
1.8 主成分分析(principal component analysis,PCA)
使用“Seurat”R软件包中的“FindVariable-Features”算法识别高度可变基因。基于高度可变基因的表达水平,使用“Seurat”R软件包中的“RunPCA”算法进行PCA,选择特征根>1的前15个主成分绘图,其中每个主成分包含15个基因。
1.9 聚类分析
用R语言进行t分布随机邻接嵌入(t-distributed stochastic neighbor embedding,t-SNE)聚类分析,获得不同细胞亚群的聚类。优化标准模块化,进行细胞聚类的可视化。识别差异表达的特征性基因,确定每个聚类的marker基因。
1.10 细胞亚群类型注释
采用“Seurat”R软件包识别各个细胞亚群中的特异性上调基因,作为对应细胞亚群的潜在标记基因。参照CellMarker数据库(http:∥xteam.xbio.top/CellMarker/)中的人类细胞标志基因或已有文献报道的细胞标志物,结合已识别的潜在标志基因,排除算法自动注释的偏倚,对细胞亚群进行注释。
1.11 细胞周期与细胞通信分析
为了预测OS细胞的细胞周期状态,基于既往文献报道的43个G1/S周期和54个G2/M周期相关的关键基因,本研究采用“CellCycleScore”函数对细胞周期进行判定。采用ggplot2(https:∥github.com/tidyverse/ggplot2)软件对细胞周期分析结果进行可视化。基于基因表达矩阵,结合已有的配体-受体信息,采用“iTALK”软件包量化配体-受体相互作用的强度,推测细胞间的互作关系。
分析流程见图1。

图1 本研究的分析流程图
2 结 果
2.1 干性指数的计算
本研究使用一类线性回归算法(one class linear regression,OCLR)获得每个OS样本的干细胞指数(mRNA expression-based stemness index,mRNAsi),实现对87个OS样本肿瘤干性的量化。87个OS患者的mRNAsi与临床指标之间(存活状态、性别、种族、是否转移、原发灶位置等)的整体关系见图2。不同存活状态(存活vs死亡)、不同性别(男性vs女性)、不同种族(白人vs亚洲人vs黑人vs未知人种)、不同转移状态(转移vs无转移),以及不同原发部位(下肢vs骨盆vs上肢)OS样本的mRNAsi之间差异均无统计学意义(P>0.05)。
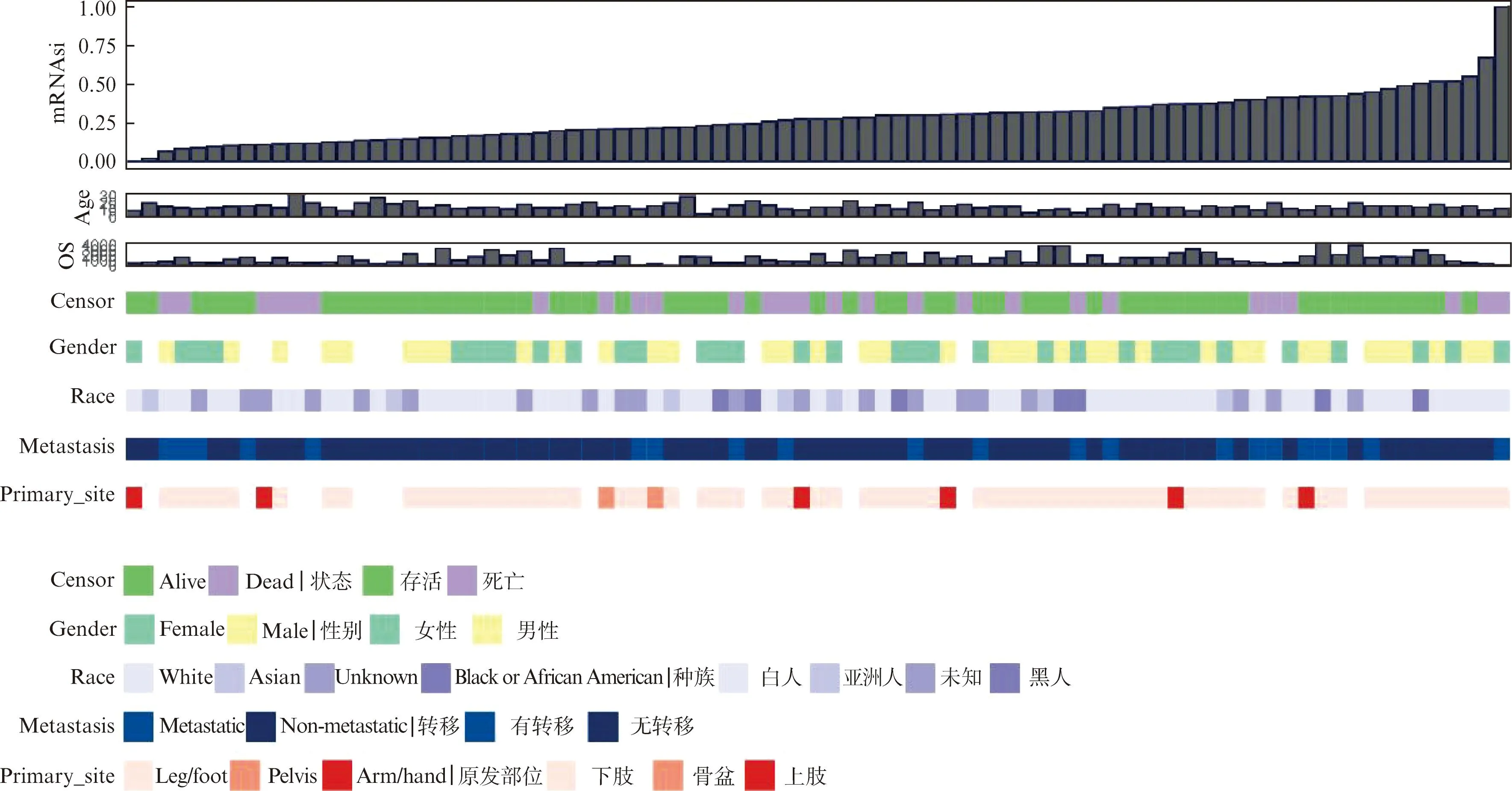
图2 骨肉瘤患者基线特征及mRNAsi水平
肿瘤干细胞(cancer stem cells,CSCs)是指拥有与正常干细胞相关特征的癌细胞,能在特定癌症样本中产生所有细胞类型。通常肿瘤干细胞被认为有形成肿瘤的潜力,特别是随着癌症的转移,能产生新的癌症病灶。mRNAsi值越高,患者肿瘤干细胞活性越高,发生转移的可能性也越大。
2.2 关键基因
韦恩图显示,皮尔逊相关性分析筛选出5 131个干性相关基因;Cox回归分析筛选出1 640个风险相关基因;KM生存分析筛选出7 066个生存相关基因。其中658个基因在3个基因集中都有统计学意义,被定义为关键基因,纳入后续分析,见图3。
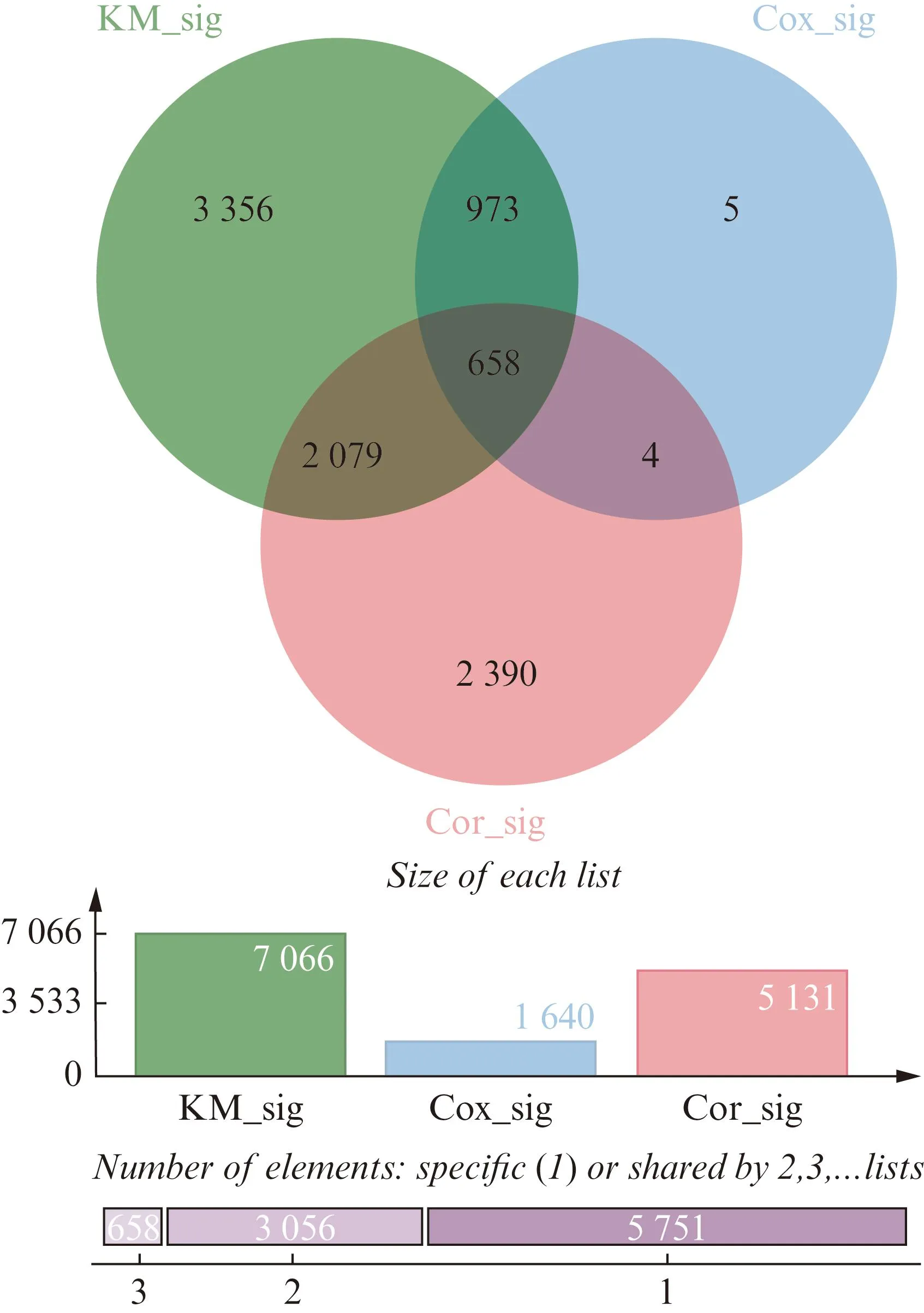
图3 关键基因的韦恩图
此外,根据骨肉瘤干性基因与mRNAsi之间的相关系数R,将R>0.6或R<-0.5作为筛选标准,进一步筛选与肿瘤干性高度相关的关键基因,最终获得了57个关键基因,其与mRNAsi之间的相关系数见图4。

图4 肿瘤干性相关基因与mRNAsi之间的相关系数
在关键基因中,NADH脱氢酶(泛醌)1β亚复合物9(NADH:Ubiquinone Oxidoreductase Subunit B9,NDUFB9)与mRNAsi之间的正相关性最强(相关系数r=0.57,P<0.001),并且NDUFB9表达水平高的OS患者预后更差(P=0.007),见图5。
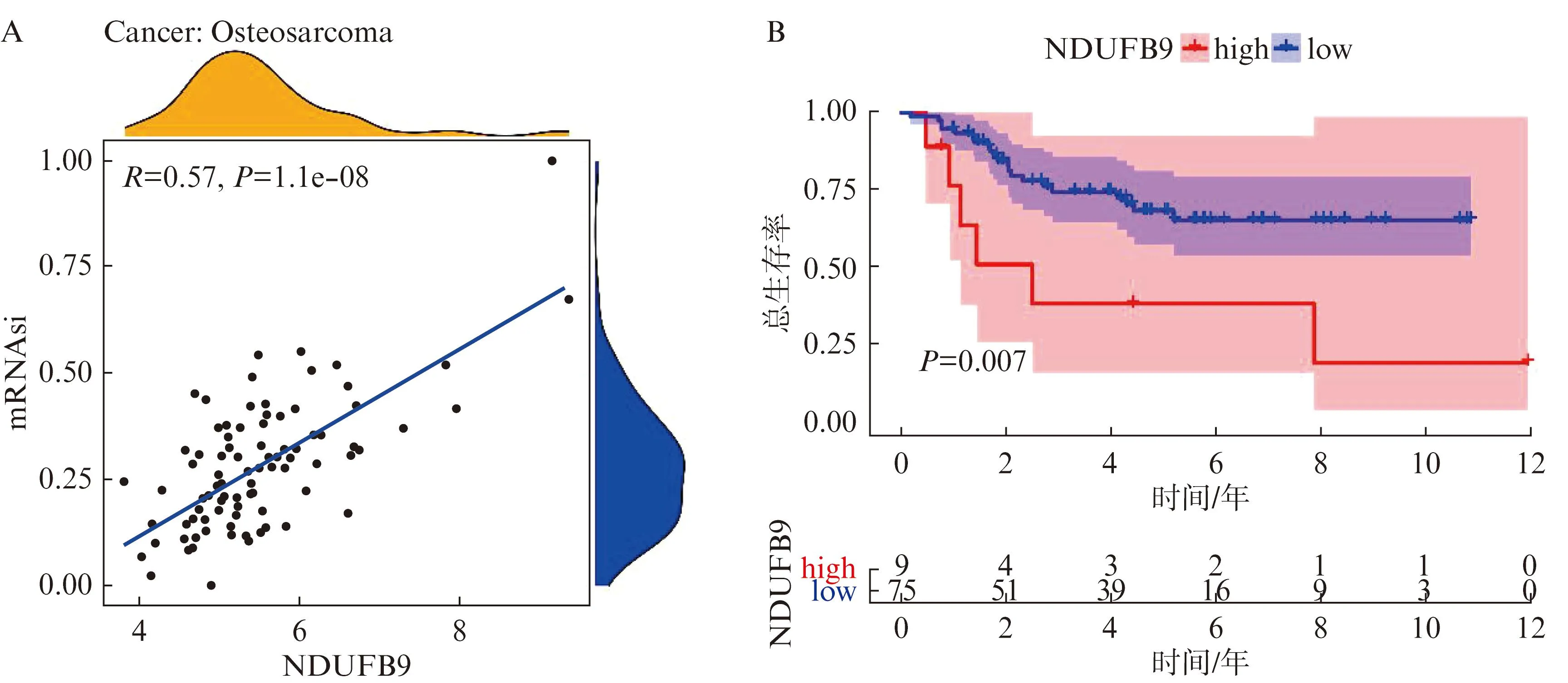
图5 NDUFB9与mRNAsi的相关性及预后价值
相反,关键基因EHD家族蛋白2(EH domain containing 2,EHD2)与mRNAsi之间的负相关性最强(相关系数r=-0.43,P<0.001),并且EHD2表达水平低的OS患者预后更差(P=0.002),见图6。
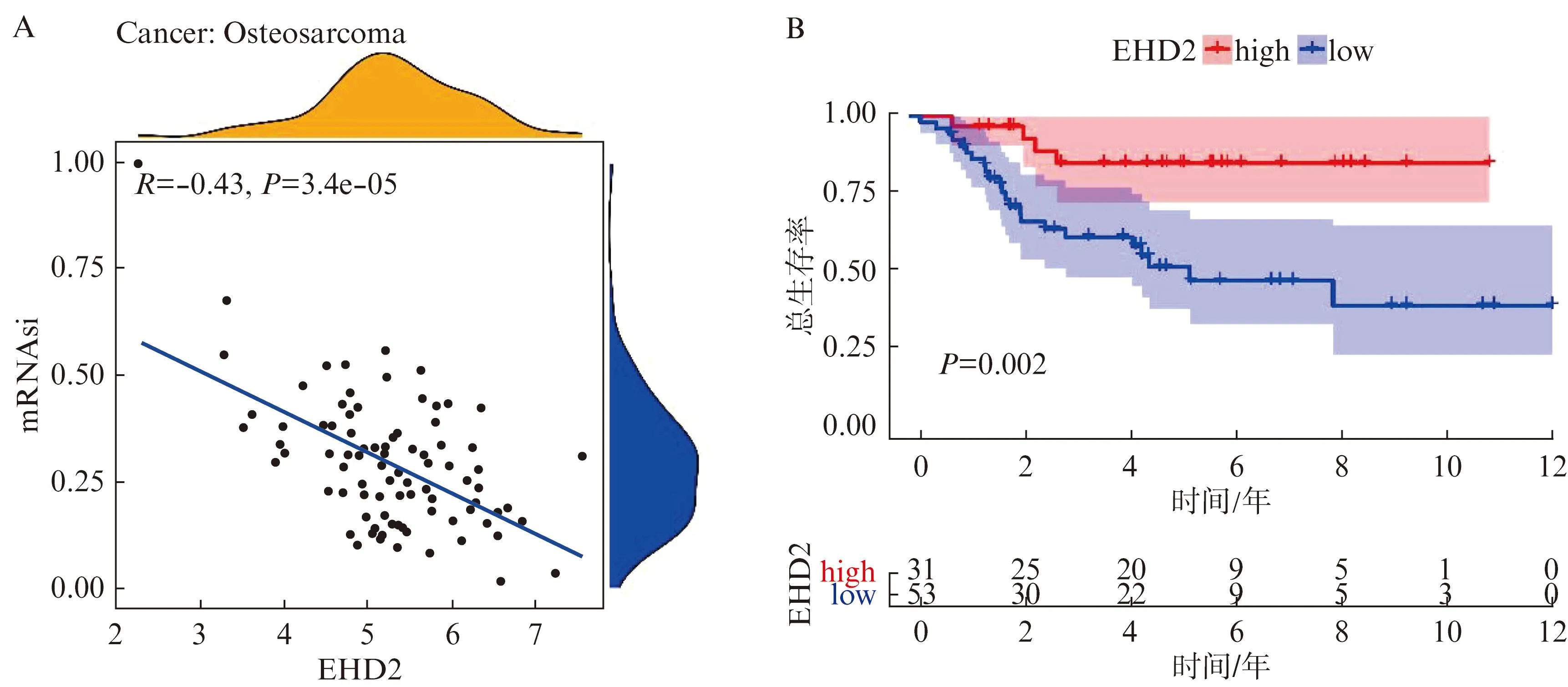
图6 EHD2与mRNAsi的相关性及预后价值
2.3 OS肿瘤组织内的主要细胞亚群及其分子特征
经过严格质控,本研究最终共获得123 307个细胞的高质量scRNA-seq数据。基于R语言,进行PCA并绘制热图,识别了15类特征根>1的主成分。各基因分组及表达差异显著,提示OS肿瘤细胞表现出显著分离趋势。在11个OS患者的肿瘤组织中,使用tSNE聚类分析将15个主成分中的细胞分为10种细胞亚群,见图7A,分别为成软骨细胞(chondroblasts)、内皮细胞(endothelial cells)、成纤维细胞(fibroblasts)、淋巴细胞(lymphocytes)、间充质干细胞(mesenchymal stem cells,MSCs)、髓系细胞(myeloid cells)、成肌细胞(myoblasts)、成骨细胞(osteoblasts)、破骨细胞(osteoclasts)和周细胞(pericytes)。病理学分析发现,大部分单细胞来源于传统骨肉瘤分型,见图7B。
上述10种细胞亚群在不同OS患者肿瘤组织中的数目和比例具有明显差异,见图7C。细胞特征图显示分化簇24(cluster of differentiation 24,CD24)主要表达于成骨细胞、间充质干细胞及成肌细胞;分化簇44(cluster of differentiation 44,CD44)主要表达于成纤维细胞和破骨细胞;MKI67主要表达于成骨细胞和间充质干细胞;分化簇133(cluster of differe-ntiation 133,CD133或Prominin 1,PROM1)主要表达于成骨细胞和周细胞;NDUFB9在多种OS肿瘤细胞亚群中表达水平上调,包括间充质干细胞和成骨细胞等;而EHD2在各肿瘤细胞亚群的表达量都较低,见图7D。CD24是一种小分子量、高度糖基化的细胞膜蛋白质,通过参与肿瘤发生发展相关信号通路(如Wnt信号通路和MAPK信号通路)调控肿瘤细胞的生长、增殖和转移,与肿瘤的侵袭转移密切相关。CD44是一种非激酶跨膜糖蛋白,可促进肿瘤细胞增殖和侵袭,在大多数肉瘤中高表达,大量研究证实其对肿瘤进展、转移和耐药性有直接影响。此外,CD44已被证实是骨肉瘤诊断和预后评价的有效标志物[19]。CD44也是检测和分离化疗抗性CSCs的特异性生物标志物[20]。MKI67表达水平反映细胞增殖状态,在癌症转移、侵袭和进展中起着至关重要的作用,是癌症的预后预测因子[21-22]。PROM1编码五跨膜糖蛋白,通常在癌细胞中过表达,通过抑制分化来维持肿瘤细胞的干细胞特性[23]。PROM1在各类肿瘤干细胞的鉴定和分选中被广泛应用。通过对OS肿瘤组织进行单细胞测序发现,本研究鉴定的关键基因NDUFB9在具有干细胞特征的细胞亚群中表达上调,进一步明确了上述基因与OS肿瘤干性特征密切相关,可作为潜在预后标志物。
为了进一步解析OS肿瘤细胞的异质性,基于CellMarker数据库中既往文献报道的细胞标志物,本研究识别并注释了10个细胞亚群,包括成骨细胞(MMP9、ACP5、CTSK)、破骨细胞(MT1X、ALPL、CYR61)、成纤维细胞(MMP13、IBSP、SPP1)、间充质干细胞(SFRP2、COMP、PLA2G2A)、髓系细胞(HLA-DRA、CCL3、CCL3L1)、周细胞(FGFBP2、COL11A1、HAPLN1)、内皮细胞(SPARCL1、RAMP2、PLVAP)、成肌细胞(SFTPC、MYLPF、PTN)、淋巴细胞(CCL5、CD69、IL32)、成软骨细胞(SSPN、COL 12A1、LRP1),见图7E。差异表达分析从10个亚群中鉴定出59 578个标志基因,各细胞亚群基因表达差异明显,见图7F。
2.4 细胞通信和细胞周期分析
克利夫兰图显示,4种经典Marker在10种细胞亚群中表达水平有明显差异。如前所述,上述10种细胞亚群在11个OS患者肿瘤组织中的比例具有显著差异,见图8A。使用“Seurat”包中的“Find-AllMarkers”函数识别不同细胞亚群的标记基因,并通过热图展示这些细胞亚群中前5个差异表达标志基因的表达水平,见图8B。细胞通信分析显示,成软骨细胞和MSCs占据了细胞通信网络的中心,主要通过配体:胶原蛋白Ⅰ型α 1链(collagen type Ⅰ Alpha 1 chain,COL1A1)和胶原蛋白Ⅰ型α 2链(collagen type Ⅰ Alpha 2 chain,COL1A2)发送信号,并通过受体低密度脂蛋白受体相关蛋白1(low-density lipoprotein receptor-related protein 1,LRP1)接收信号,见图8C、D。细胞周期分析显示淋巴细胞、MSCs和成骨细胞表现为细胞增殖较为活跃,主要处于G2/M期;而周细胞和成肌细胞主要处于相对静息的S期,见图8E。而NDUFB9在MSCs和成骨细胞等增殖较为活跃的细胞亚群中的表达水平较高,并且MSCs在OS肿瘤微环境细胞间通讯中占据着中心枢纽地位,这进一步验证了NDUFB9高表达与肿瘤细胞干性之间的潜在关联及其在促进肿瘤发生发展中的潜在作用。此外,EHD2在上述增殖活跃的肿瘤细胞亚群中表达量极低,与之前的结果一致,即EHD2与肿瘤细胞干性之间呈负相关关联。

图8 骨肉瘤细胞亚型识别、细胞通信与细胞周期分析
基于单细胞水平进一步分析先前识别的57个差异表达干性相关基因(R>0.6或R<-0.5)是否在不同细胞亚群间仍存在差异表达。结果显示,与肿瘤干性呈负相关关系的20个基因中,共有18个基因表现出明显差异表达的特征,并且这些基因在淋巴细胞、MSCs和成骨细胞等增殖活跃的细胞亚型中表达水平极低,见图9。此外,与肿瘤干性呈正相关关系的37个基因中,共有25个基因表现出明显差异表达的特征,并且这些基因在淋巴细胞、MSCs和成骨细胞等增殖活跃的细胞亚型中普遍高表达,且表现出一定的共表达趋势,这进一步验证了这些基因与OS肿瘤干性之间的潜在关联,见图10。

图9 单细胞测序识别的差异表达干性相关基因(与mRNAsi呈负相关关系)的细胞特征图
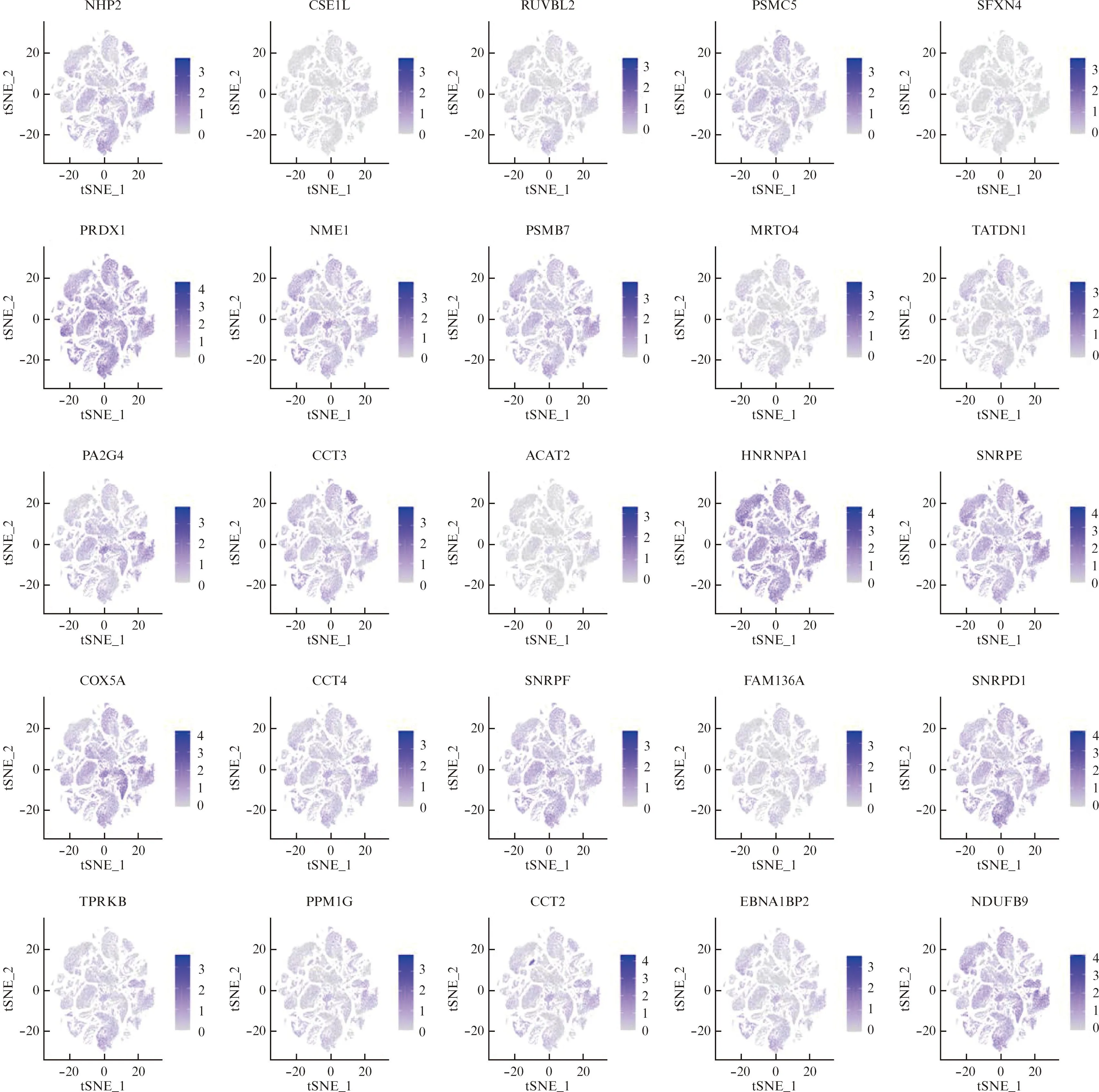
图10 单细胞测序识别的差异表达干性相关基因(与mRNAsi呈正相关关系)的细胞特征图
3 讨 论
CSCs是参与肿瘤侵袭转移及耐药的关键细胞亚群[24]。肿瘤细胞分化程度越低,则其干细胞样特征越显著,肿瘤恶性程度也越高。肿瘤干性的本质是多基因表达异常介导下游信号通路失调,通过维持CSCs的自我更新或诱导肿瘤细胞去分化而获得干细胞样特征。然而,目前OS干性特征的发生机制尚未明确。本研究通过TARGET数据库联合机器学习描绘了OS干性特征,并挖掘了调控OS肿瘤干性特征的潜在机制。结果发现,mRNAsi与OS患者的原发部位、生存状态和肿瘤转移相关,是影响OS患者预后的不利因素。
基于OS患者RNA-seq数据,本研究共筛选出658个关键基因。这些基因不仅与肿瘤干性显著相关,而且与OS患者预后及生存显著相关,是OS干细胞靶向治疗的潜在靶点。在关键基因中,NDUFB9和EHD2与mRNAsi之间的相关性最强,且均与OS患者的总生存期显著相关。
NDUFB9编码线粒体膜呼吸链NADH脱氢酶(复合物Ⅰ)的一个辅助亚单位[25]。NDUFB9突变可导致复合物Ⅰ缺乏[26]。复合物Ⅰ可以氧化NADH,减少泛醌,并通过线粒体内膜运输质子,从而产生质子动力。众所周知,线粒体和能量代谢的改变与癌变之间存在着紧密关联。癌细胞会偏向以无氧糖酵解取代正常的有氧循环进行能量供给,所以癌细胞线粒体的生物氧化方式与正常细胞有显著区别,这一现象被称为瓦氏效应。瓦氏效应促进了肿瘤细胞适应低氧和快速增殖[27]。多项研究证明上调NDUFB9可促进肿瘤转移[28-29]。最近有研究基于单细胞测序分析和转录组分析发现NDUFB9在子宫内膜癌组织中显著上调,在子宫内膜癌的发生发展中发挥着重要作用[30]。此外,NDUFB9表达异常引起的复合物Ⅰ活性改变增强了癌细胞的侵袭性,导致患者的不良预后[29]。
EHD2编码Eps15同源结构域,是一种肿瘤抑制基因,在多种肿瘤中表达水平明显降低[31]。EHD2表达与组织学分级、淋巴结转移和肿瘤大小有关。此外,EHD2的过度表达抑制癌细胞的迁移和侵袭,而敲除EHD2则促进癌细胞的迁移和侵袭[31]。最新研究表明,EHD2与多种癌症显著相关。与正常组织相比,食管鳞状细胞癌中EHD2蛋白水平明显降低,并且EHD2的低表达促进了食管鳞状细胞癌细胞的迁移[32]。在卵巢癌、恶性黑色素瘤和肝细胞癌中也发现了EHD2的明显低表达[33-35],而低表达EHD2的患者生存期更短,预后更差。
肿瘤异质性包括瘤内异质性和瘤间异质性,源于形态学和表观遗传学的可塑性,也可归因于基因表达谱不同的肿瘤细胞克隆共存[36]。单细胞测序在探究肿瘤异质性方面具有独特优势,特别是在OS这种具有高度异质性、难以用整体测序方法进行描述的复杂肿瘤中。
本研究绘制了OS患者的转录组学单细胞图谱,并探究了复杂的OS肿瘤微环境以及肿瘤异质性。通过对11个OS患者的scRNA-seq数据进行质控、降维后,识别并注释了10种不同的细胞亚群,提示OS肿瘤细胞之间存在异质性。在通过质控的123 307个单细胞中,成骨细胞比例最高,其次是髓系细胞和成纤维细胞。在复杂的OS肿瘤微环境中,细胞之间存在着广泛的细胞通信。本研究使用“iTALK”R软件包来分析不同细胞亚群间的受体配体对,从而预测细胞间通讯模式。成软骨细胞和MSCs占据了通讯网络中心,主要通过配体COL1A1和COL1A2发送信号,并通过受体LRP1接收信号,在该通讯网络中发挥重要作用。COL1A1与COL1A2编码产物是Ⅰ型胶原蛋白的重要组成成分,可以形成Ⅰ型胶原蛋白的三螺旋结构[37]。Ⅰ型胶原蛋白是骨骼中胶原蛋白形成所必需的,并且它也参与细胞外基质(extra cellular matrix,ECM)的合成和细胞形态改变。研究表明,COL1A1和COL1A2在多种肿瘤组织中的表达比在正常组织中更高,并且都与肿瘤细胞的侵袭和增殖有关[38-39]。重要的是,本研究鉴定的关键基因NDUFB9在多种细胞亚群中表达水平都较高,包括间充质干细胞和成骨细胞等;而EHD2在各肿瘤细胞亚群的表达量极低或不表达。NDUFB9与多种特异性干性标志物存在共表达趋势,进一步明确了NDUFB9与OS肿瘤细胞干性的密切联系。相反,EHD2在各类OS细胞亚群中低表达或几乎不表达。结合细胞周期分析结果发现,分裂增殖旺盛的细胞亚群(如间充质干细胞和成骨细胞)高表达NDUFB9,低表达EHD2。基于单细胞测序,可更加深入地认识OS肿瘤细胞的分子特征,为剖析其肿瘤微环境、探索肿瘤异质性、寻找诊断和治疗靶点提供理论依据。
总之,本研究通过整合生物信息学分析筛选了OS预后相关的关键干性基因,为靶向药物研究提供了重要的分子基础。此外,基于OS患者scRNA-seq数据,本研究描绘了OS单细胞转录组图谱,初步揭示了OS肿瘤异质性,分析了潜在的细胞通信模式,从单细胞水平验证了关键基因在OS肿瘤细胞中的表达水平。
致谢:特别感谢上海格致中学(奉贤校区)的汤慧婷同学在论文参考文献整理中的贡献。
