Next-generation Sequencing of MHC Class I Genes Reveals Trans-species Polymorphism in Eutropis multifasciata and Other Species of Scincidae
2024-01-02ShufangZHANGYoufuLINYingzhiCHENGHaiyunYANGXiamingZHUYuDULonghuiLINYanfuQULianCHENandHongLI
Shufang ZHANG ,Youfu LIN ,Yingzhi CHENG ,Haiyun YANG ,Xiaming ZHU ,Yu DU,3 ,Longhui LIN,Yanfu QU,Lian CHEN* and Hong LI*
1 College of Life Sciences,Nanjing Normal University,Nanjing 210023,Jiangsu,China
2 College of Life Sciences,Nanjing Forestry University,Nanjing 210037,Jiangsu,China
3 Hainan Key Laboratory of Herpetological Research,College of Fisheries and Life Science,Hainan Tropical Ocean University,Sanya 572022,Hainan,China
4 Herpetological Research Center,College of Life and Environmental Sciences,Hangzhou Normal University,Hangzhou 311121,Zhejiang,China
Abstract The genes of the major histocompatibility complex(MHC)encode cell surface proteins that are essential for adaptive immunity.MHC genes show the most prominent genetic diversity in vertebrates,reflecting the adaptation of populations to their evolving environment,population survival and reproduction.In the present study,we used nextgeneration sequencing (NGS) to study the loci polymorphism of exon 3 of the MHC class I genes in an ovoviviparous skink,the many-lined sun skink,Eutropis multifasciata and five other species of Scincidae,to quantify genetic variation.In addition,we genotyped the same MHC class I genes of E.multifasciata using clone sequencing,to directly compare the effectiveness of both analytical techniques for MHC genotyping.NGS detected 20 MHC class I alleles in E. multifasciata,and 2 to 15 alleles in the other five Scincidae species.However,clone sequencing detected only 15 of those MHC class I alleles in E. multifasciata.In addition,transspecies polymorphism of MHC class I genes was studied by constructing a phylogenetic tree using the gene sequences obtained by NGS.Phylogenetic analysis revealed that MHC class I alleles were shared among different species of Scincidae with trans-species polymorphism,and did not exhibit specific genealogical inheritance.These results have important implications for understanding polymorphism interspecies diversity in the MHC genes of Scincidae,and the evolution of the MHC more broadly.
Keywords Eutropis multifasciata,major histocompatibility complex,next-generation sequencing,Scincidae,trans-species polymorphism
1.Introduction
The immune system is a complex network of organs,cells and proteins which play vital roles in protecting an organism from pathogens (Elbers and Taylor,2016).The major histocompatibility complex(MHC)is part of the sophisticated“adaptive immune system”;it encodes the molecules that are responsible for antigen recognition,and subsequent binding(Sommer,2005).The MHC is divided into three classes(MHC class I,II and III) based on the location and function of the genes within it.MHC class I genes primarily encode regions which identify and respond to polypeptides of endogenous antigens (i.e.,antigens that have been newly synthesized in antigen-presenting cells)and initiate the subsequent immune response (Neefjeset al.,2011).In order to bind to different antigenic peptides,the antigen-binding regions(exons 2 and 3)of MHC class I must be highly polymorphic.Polymorphisms in MHC class I genes have been studied across the Cercopithecinae (Caskeyet al.,2019),Lacertidae (Yuanet al.,2014) and Muscicapidae (Biedrzyckaet al.,2017),generally using exon 3 as the typing target.Perhaps not surprisingly,individuals with high polymorphisms in MHC class I genes have stronger immune function in response to a wide range of pathogens(Radwanet al.,2020).
Within the jawed vertebrate genome,the evolutionary history of the MHC has varied across clades,resulting in the MHC consisting of a highly polymorphic,multigene family.Polymorphisms of MHC genes are created by individual adaptation in a population,resulting in the occurrence of multiple alleles for a single gene locus.As such,MHC genes are important markers for species adaptations,like developing resistance to parasites(Hackinget al.,2018)or acclimating to different geographical locations(Sagonaset al.,2019).
Another form of polymorphism in MHC genes is transspecies polymorphism (TSP),which is the occurrence of identical or similar alleles among closely related species,that have not resulted from convergent evolution or gradual crossfertilization (Kleinet al.,2007).In general,MHC genes are homologous among species,have high sequence polymorphism,and linkage disequilibrium among different loci.For example,using exon 3 of the MHC class I genes researchers reconstructed the phylogenetic tree of three
Eremiasskink species (E.przewalskii,E.brenchleyi,andE.multiocellata).The resulting tree showed that the MHC class I gene lineages were not clustered by species,rather,they exhibited TSP across the three species (Yuanet al.,2014).In such cases,MHC alleles could be older than the species and would thus have sufficient time to accumulate multiple interallelic variations,resulting in the development of the TSP(Kleinet al.,2007).
The genes characteristics of the MHC are well described in mammals(especially humans and mice),birds,amphibians and fish,but little is known about their structural features in reptiles(Arnold and Hämmerling,1991;Jongsmaet al.,2019),leaving a large phylogenetic gap.To understand the wider evolution of the MHC,we must study the sequence characteristics of reptilian MHC genes.The reptilian family Scincidae is large and diverse,both morphologically and ecologically.Because of this,the Scincidae provide an ideal model system to obtain fundamental information about the evolution of MHC diversity and TSP across the wider family.The many-lined sun skink(Eutropis multifasciata)is a mediumsized,tropical lizard,native to China,India,Bangladesh and the Philippines (Duet al.,2012).It is ovoviviparous and a suitable species to investigate the proximate and ultimate reasons for variation in the reproductive patterns of reptiles(Sunet al.,2012).Populations of this skink are expected to decline dramatically in the future,due to the medicinal and culinary values of this species(Dunget al.,2011).In 2023,the State Forestry Administration of the People’s Republic of China recognized this threat and identified thatE.multifasciatawas of important economic and scientific value and recommended state protection as within China.Given this context,the study of the MHC inE.multifasciataalso provides information for the conservation of endangered species.
When sequencing genes,researchers generally employ a protocol using the polymerase chain reaction(PCR)to amplify the corresponding alleles.Initially,the cloning and then sequencing technique uses the plasmid incompatibility principle to isolate heterozygous alleles and obtain haplotypic gene information,commonly used for the confirmation of new alleles,obtaining gene sequences and polymorphism analyses (Christet al.,1994).However,this may not be the most suitable method for sequencing MHC genes.Previous studies have found that in many species sequencing MHC genes with PCR can cause gene duplication,copy number variation and co-amplification of alleles from different loci,making the determination of individual genotypes impossible(Babik,2010).Fortunately,next-generation sequencing(NGS)is a massively parallel sequencing technology with high throughput,scalability and speed.This technology facilitates the accuracy and reproducibility of genotyping polymorphic gene families,such as the MHC,in non-model organisms(Groganet al.,2016;Shaheenuzzamnet al.,2020).
In the present study,we use NGS to investigate exon 3 of the MHC class I genes in six skink species(E.multifasciata,P.chinensis,S.modesta,S.reevesii,S.indicusandS.incognitus).MHC genotyping is difficult due to the high degree of polymorphism between MHC gene sequences and the variation in allele numbers between species (Rekdalet al.,2018).In order to compare the ability of different techniques to detect MHC genes,we also used clone sequencing to detect exon 3 of the MHC class I genes inE.multifasciata.The results of this study have important implications for understanding polymorphism and interspecific diversity in the MHC genes of the Scincidae.
2.Materials and Methods
2.1.Sample collection and DNA extractionWe collected 14E.multifasciataand 8 other lizards in China(Table 1).The samples were kept in the Reptile Laboratory at Nanjing Normal University.DNA was extracted from the muscle tissues of 22 lizard samples using the DNeasy Tissue Kit(Qiagen,Germany)according to the manufacturer’s protocol.

Table 1 Sample information of six species of Scincidae.
2.2.MHC genotyping by an amplicon-based NGS approachThe universal primers(Murphyet al.,2009)2MHCF(5'-CAG CAG ATG TAT GGC TGT GA-3') and 2MHCR (5'-GCAGAT CTC CTC CAG GTA G-3')were used to amplify exon 3 of the MHC class I genes(corresponding to the α-2 region)in six species(E.multifasciata,Plestiodon chinensis,Scincella modesta,S.reevesii,S.indicus,andS.incognitus)and a unique barcode was added to the 5' end of the primers to identify different individuals.The PCR amplification reaction system was 50 μL,including 10 μL Premix Taq,1 μL of each primer(10 μmol/L),2 μL template DNA(100 ng)and ddH2O.The following cycle conditions were used for PCR amplification:94°C for 5 min;35 cycles of 94°C for 30 s,60°C for 30 s and 72°C for 1 min;72°C for 10 min.PCR products were mixed and purified with the QIAquick Gel Extraction Kit(Qiagen,Germany).The size and quality of the pooled amplicons were verified using the TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina,USA)on the 2100 Bioanalyzer(Agilent,USA).The library was then sequenced on the Illumina NovaSeq and 250 bp pairedend sequences were obtained (Novogene Bioinformatics Technology,China).
The raw sequencing data contained a variety of error messages that interfered with the subsequent analysis,such as sequencing junctions,low-quality bases and undetected bases(expressed as N).Therefore,the data were filtered by four criteria: (1) filtering out splice sequences and barcode sequences from the reads;(2) splicing pairs of reads with overlaps using FLASH VI.2.7(Magočet al.,2011);(3)filtering the spliced data using QIIME(Caporasoet al.,2010)to remove chimeric sequences with high N or low quality;and (4)filtering out chimeric sequences from the spliced sequences.The Illumina data were then clustered using Vsearch(Rogneset al.,2016) and amplicons with a frequency of less than 1%were discarded.We only considered sequences that were present in more than one individual to be alleles(Zhaoet al.,2013).
2.3.MHC genotyping by clone sequencingUniversal primers 2MHCF and 2MHCR were used to amplify exon 3 of MHC class I genes inE.multifasciata.PCR reaction procedures and systems were as described above.Amplified PCR products were then separated using agarose gel electrophoresis,and the amplified bands of 14 individuals ofE.multifasciatawere excised and cleaned (MiniElut RNA-Pure Kit,Genebase Biogene Technology).The purified PCR product was ligated to the pMD19-T vector(TaKaRa,Japan)and transformed into DH5α receptor cells.PCRs were then performed for positive clones.Each individual then had 10 colonies and the bacterial solution containing the target fragment was sent to Shanghai Sangon Biological Engineering Technology &Services Co.,Ltd.for sequencing.
The initial quality and length filtering of raw data was performed with ChromasPro (v 2.1) to output FASTA files.The target sequences were obtained by removing redundant sequences using the MEGA X (Kumaret al.,2018).The sequences obtained from clone sequencing were imported into NCBI (https://www.ncbi.nlm.nih.gov/) for BLAST to identify MHC class I genes.The sequences were imported into MEGA X and aligned using Clustal W (Larkinet al.,2007).Haplotype information for MHC sequences was obtained by DnaSP 5.0.MHC class I alleles were named with reference to the naming principle:a new allele needed to be identified by its occurrence in at least two clones of the same individual or in different individuals (Kennedyet al.,2002) and named sequentially according to the frequency of allele occurrence.
2.4.Phylogenetic analysisThe sequences from clone and next-generation sequencing were imported into DnaSP 5.0 to calculate the nucleotide diversity (π) and variable sites (S) of MHC class I genes.The TCS Network algorithm in Popart(Hayeset al.,2022) was used to construct separate network relationships between the diverse haplotypes of Scincidae.The exon 3 sequences of MHC class I derived from 6 species in the present study combined with other 4 species (Ctenophorus decresii,Eremias multiocellata,E.przewalskii,andE.brenchleyi)were used for phylogenetic analysis(Supplementary Table S1 for GenBank accession numbers).The sequences were aligned using MAFFT implemented in PhyloSuite(Zhanget al.,2020).The phylogenetic tree was created by the maximumlikelihood (ML) method implemented in IQ-TREE with a bootstrap of 1 000 replicates.The phylogenetic tree was visualized with the iTOL online tool(https://itol.embl.de/).
3.Results
In this study,the exon 3 sequences of MHC class I genes were successfully amplified in 22 samples fromE.multifasciata,P.chinensis,S.modesta,S.reevesii,S.indicus,andS.incognitus.
3.1.Comparison of clone sequencing and NGSThe nucleotide sequences of all alleles identified by sequencing did not reveal a stop codon,indicating that these alleles all encode functional proteins.A total of 20 alleles were identified in 14 individuals.According to the naming principle of MHC class I gene sequences,20 MHC class I sequences were namedEumu-UA*01toEumu-UA*20(Table 2).Fifteen alleles were identified by clone sequencing and all of these alleles were also detected by NGS.Furthermore,clone sequencing identified samples with 3 to 7 alleles,with half of the samples having 3 alleles and only one sample having 7 alleles.The number of alleles detected by NGS ranged from 4 to 14,with four samples containing nine alleles,five samples containing more than ten alleles,and five samples containing fewer than nine alleles(Supplementary Table S2).
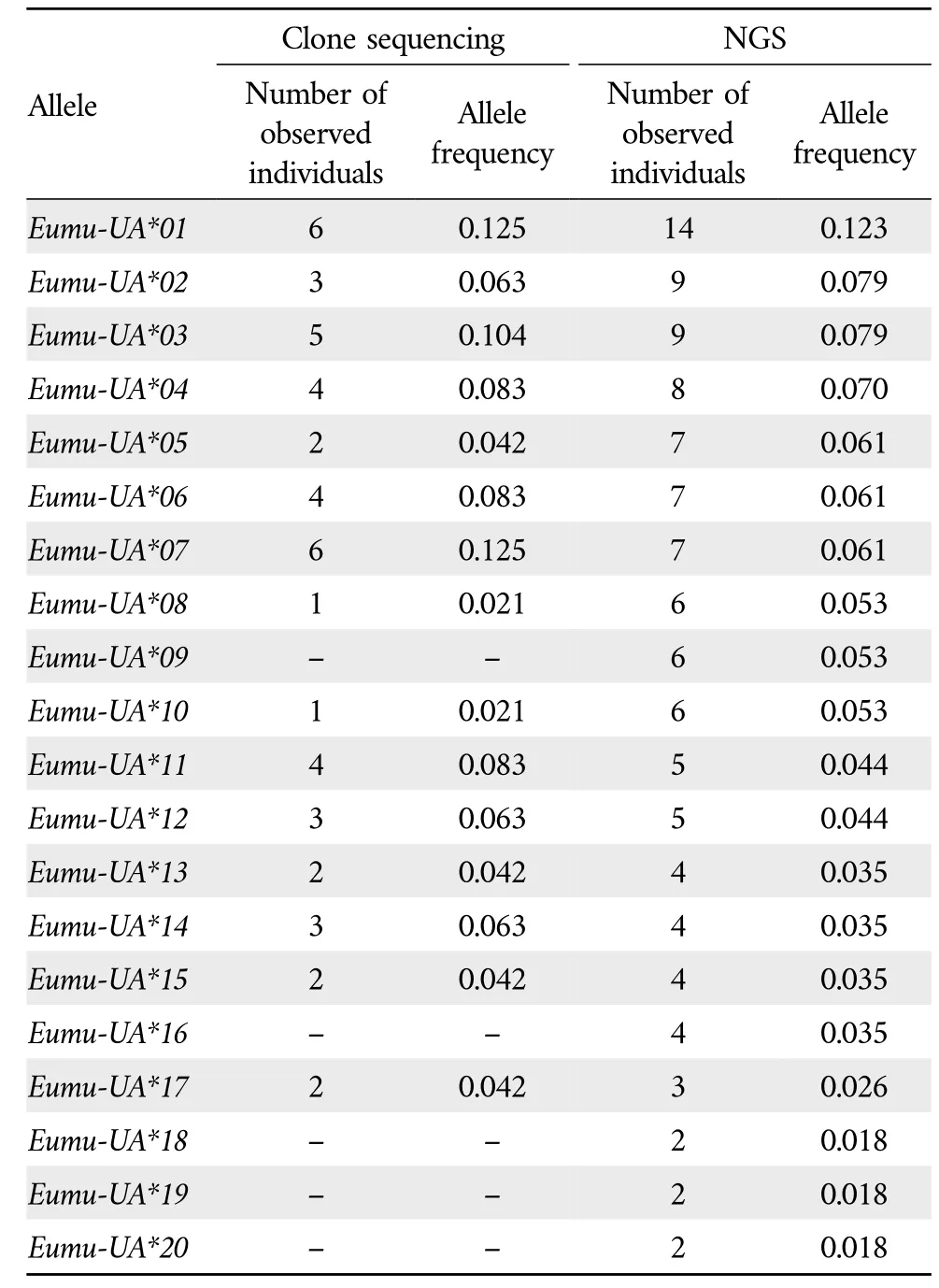
Table 2 Allele frequency of MHC I gene exon 3 of E.multifasciata based on clone sequencing and NGS.
3.2.Class MHC class I genes sequence polymorphismThere were some differences in nucleotide diversity among different species.The number of variable sites detected forE.multifasciatawas 90,with a nucleotide diversity of 0.101.No singleton variable sites were presented,but 90 parsimony informative sites were detected(Table 3).
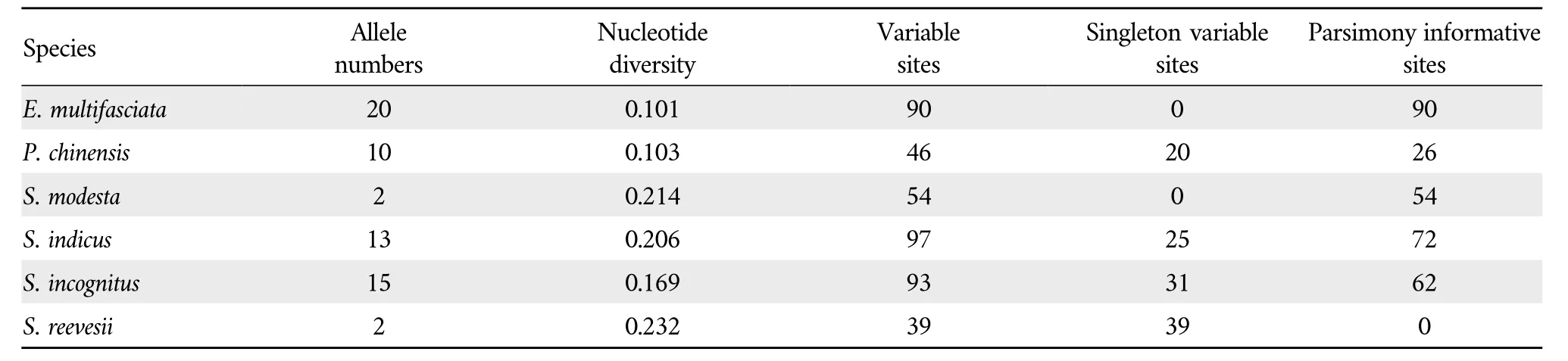
Table 3 Genetic diversity of MHC class I genes in 6 species of Scincidae.
In the other five Scincidae species,the number of variable sites ranged from 39 to 97,and the nucleotide diversity was 0.103-0.232.There were no singleton variable sites in exon 3 of the MHC class I genes inS.modesta,and the value of the parsimony informative sites was 54.No parsimony informative sites were present inS.reevesii,and the value of the singleton variable sites was 39.Three skinks(P.chinensis,S.indicus,andS.incognitus)had both singleton variable sites and parsimony informative sites.A total of 15 alleles were present inS.incognitus,13 alleles were present inS.indicusand 10 alleles were present inP.chinensis.S.modestaandS.reevesiieach contained at least two allelic loci(Table 3).
3.3.MHC class I genes trans-species polymorphismThere were eight alleles(Eumu-UA*01,Eumu-UA*02,Eumu-UA*05,Eumu-UA*06,Eumu-UA*07,Eumu-UA*08,Eumu-UA*11,andEumu-UA *14) shared with four species (E.multifasciata,S.indicus,S.incognitus,andP.chinensis).E.multifasciataandS.indicusshared 13 alleles.E.multifasciataandP.chinensisshared 10 alleles.No shared alleles were found amongE.multifasciata,S.modesta,andS.reevesii.TheE.multifasciatahad five unique alleles(Eumu-UA*09,Eumu-UA*10,Eumu-UA*15,Eumu-UA*16,andEumu-UA*18),and theS.indicusonly had one alleleSpinc-UA*01(Figure 1).
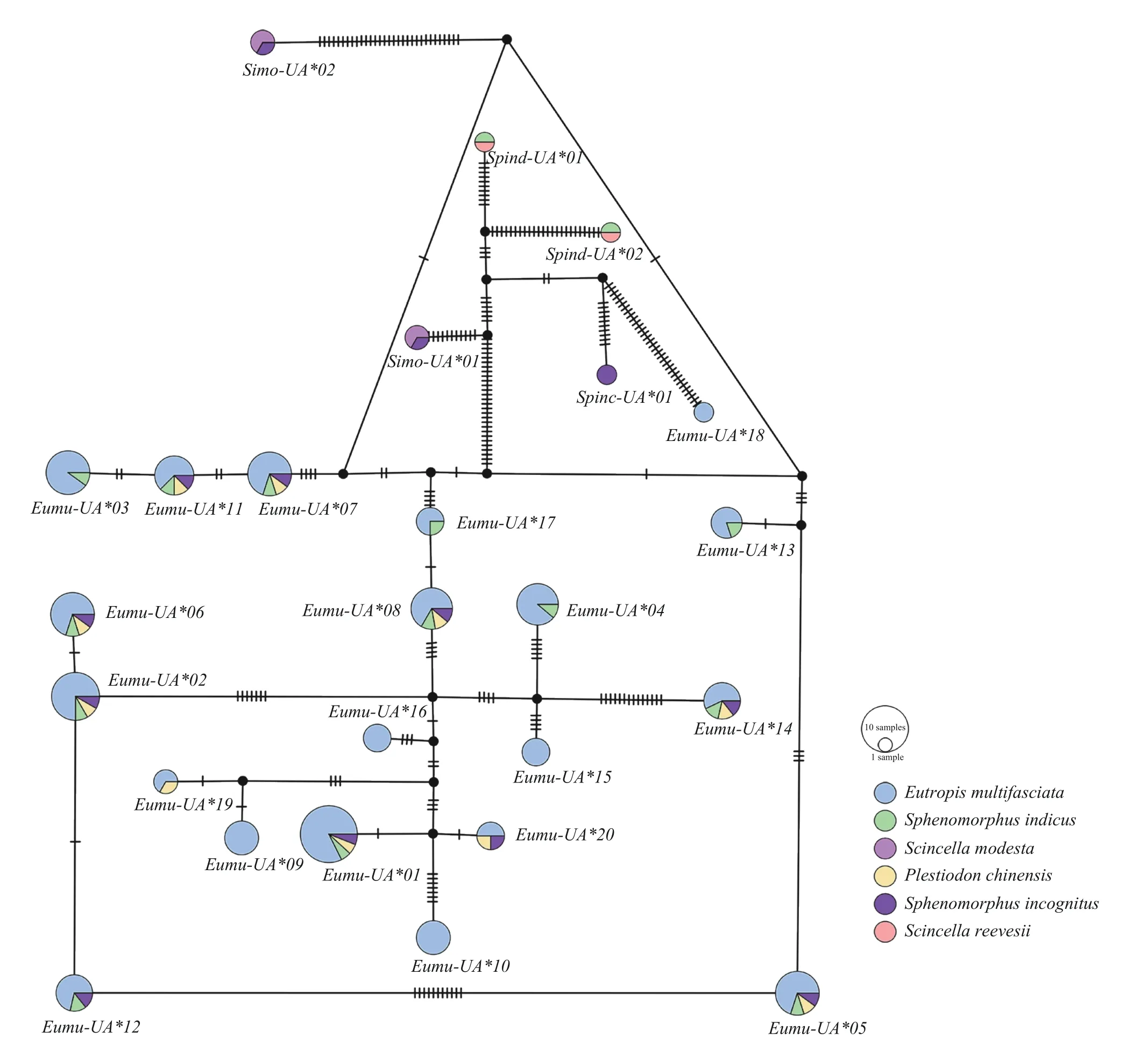
Figure 1 Median-joining network of MHC class I exon 3 alleles found in Scincidae.Each circle represents a haplotype,and the size of the circle indicates the number of samples contained in that haplotype.The line between two circles indicates that the two haplotypes are related,and the short line on the line indicates the number of nucleobase substitutions required to change from one haplotype to another.
The evolutionary relationship of MHC class I was explored by constructing a phylogenetic tree of the sequences of exon 3 from ten lizard species(Figure 2).It was found that the MHC class I exon 3 sequences of theEremiaslizards(E.multiocellata,E.przewalskii,andE.brenchleyi) clustered into one large branch,while those ofC.decresiiclustered into a single branch.However,the MHC class I exon 3 sequences of the six lizards in the family Scincidae were not clustered into one branch according to the family.Although the phylogenetic tree formed sub-branches of Scincidae,Lacertidae and Agamidae,it did not show obvious phylogenetic characteristics,and there was TSP in the exon 3 sequence of MHC class I genes.
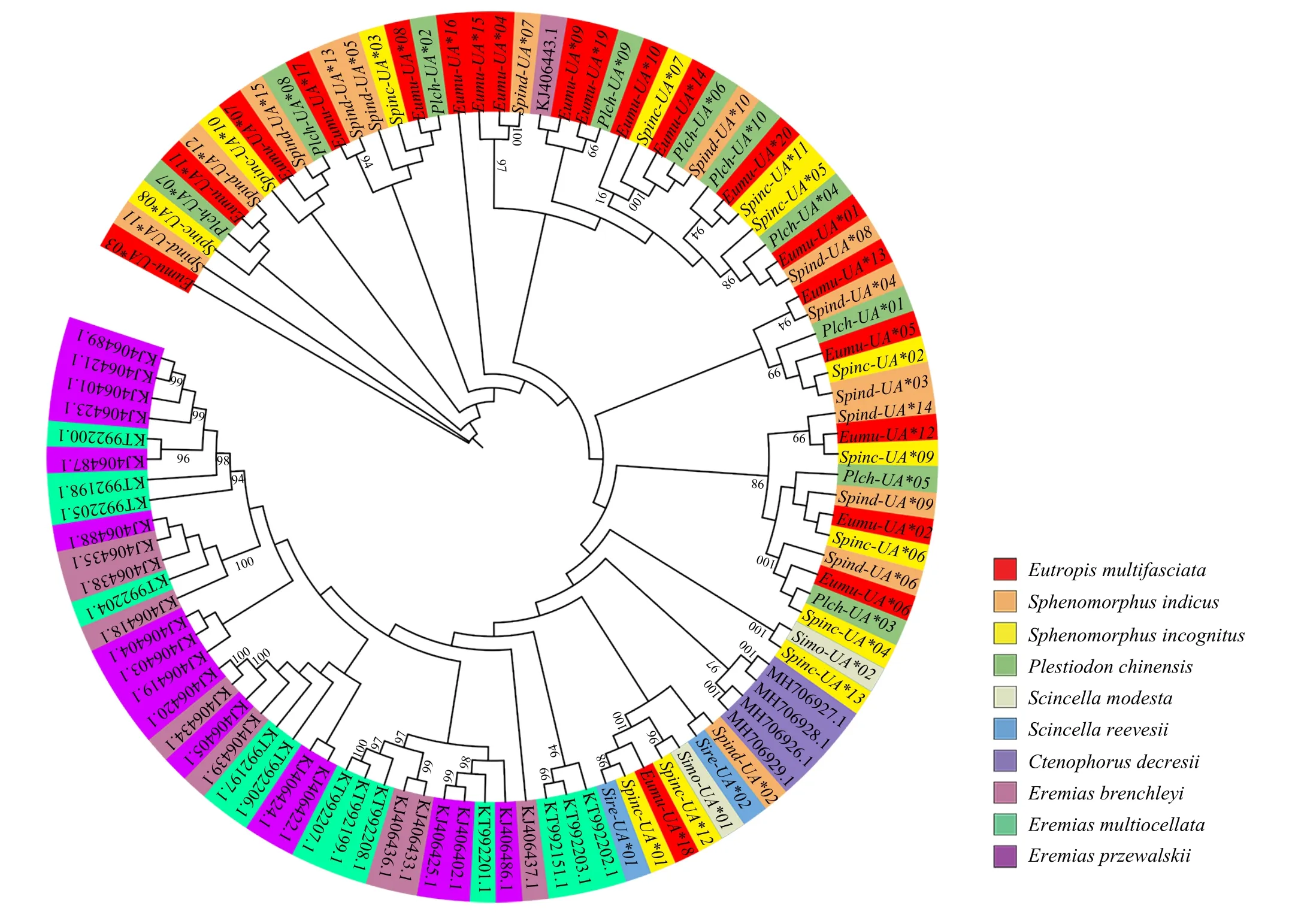
Figure 2 The Maximum Likelihood (ML) phylogenetic tree based on the MHC class I alleles from Eutropis multifasciata, Plestiodon chinensis,Scincella modesta, Scincella reevesii, Sphenomorphus indicus, Sphenomorphus incognitus, Eremias multiocellata, Eremias przewalskii, Eremias brenchleyi,and Ctenophorus decresii.
4.Discussion
4.1.Characteristics of MHC class I genes in E.multifasciataThe MHC plays an important role in the immune response of vertebrates.The inherent level of genetic variation and function within a species’ MHC underlies disease resistance,directly affecting the survival of individuals and even the fitness of entire populations(Miniaset al.,2016).Additionally,the level of genetic variation in MHC genes can be an important indicator for evaluating the evolutionary potential of adaptation in endangered wildlife(Akiyamaet al.,2017).Genetic diversity within the MHC is reflected in allelic and nucleotide diversity,as well as in the number of MHC genes.In the present study,amplicon-based NGS yielded a total of 20 MHC class I alleles inE.multifasciata.In comparison with other reptile families,the number of MHC class I alleles in the ornate dragon lizard(C.ornatus)was close to that ofE.multifasciata,ranging from 3-14 (Radwanet al.,2014).In contrast,the Tuatara(Sphenodon spp.)possessed 49 MHC class I alleles(Milleret al.,2010),which was significantly higher than theE.multifasciata.However,only six MHC class I alleles were found in a more closely related skink speciesC.decresii(Yuanet al.,2014),which was lower than that ofE.multifasciatain the present study.The main driver of genetic diversity within the MHC is pathogen-mediated selection,but other evolutionary processes(e.g.,genetic drift and gradual infiltration)may also increase genetic diversity within the MHC (Sagonaset al.,2019).
4.2.Comparison of MHC typing using clone sequencing and next-generation sequencingTo understand the functional significance of allelic diversity in the MHC within the context of evolutionary ecology,pathogen resistance and conservation,it is imperative that the MHC composition of individuals is correctly assessed.Several methods have been used to type MHC genes,such as denaturing gradient gel electrophoresis (DGGE);singlestrand conformation polymorphism (SSCP);reference strand-mediated conformational analysis (RSCA);and direct sequencing after PCR purification (Lenzet al.,2008).The most widely used of these methods are clone sequencing and next-generation sequencing,which have their own advantages and limitations.The number of classical MHC genes may vary intra-or inter-species(de Grootet al.,2015;Plasilet al.,2022).The traditional method is to amplify individual samples using primers and pool the amplification products from the individual samples for sequencing.However,the MHC genes of many species exhibit gene duplications,copy number variations,and common alleles from different loci,making it impossible to determine individual genotypes using traditional methods (Babik,2010).In addition,clone sequencing can only be performed on small batches of samples,which is cumbersome and less sensitive,i.e.,susceptible to allelic deletions.Clone sequencing is also limited by the size of the cloning vector,the larger the amplified fragment,the more difficult it is to clone.NGS technology is easy to use and powerful,it detects large samples of MHC alleles concurrently during typing.However,it is difficult to obtain complete sequences and usually targets one or more exons(Chenget al.,2022).
In the present study,the sequences of exon 3 of the MHC class I genes ofE.multifasciatawere sequenced using conventional clone and next-generation sequencing techniques,respectively.A total of 15 MHC alleles were detected by both clone sequencing,and NGS techniques,however NGS also detected an additional five new MHC alleles.This demonstrates that NGS is superior for sequencing highly polymorphic MHC genes,and can thus reduce the risk of allelic loss caused by preferential amplification of fewer alleles.For example,Eumu-UA*01was detected in only 6 individuals by clone sequencing,whereas it was detected in all 14 individuals by NGS.Therefore,NGS technology has clear advantages over traditional clone sequencing for genotyping of the MHC.This builds on previous studies utilizing NGS for MHC in other taxa.Genotyping of the MHC in Korean native chickens using long-range PCR in combination with NGS confirmed that NGS technology could be used effectively to detect MHC variants in avian fauna(Ediriweeraet al.,2022).Another study utilizing NGS to genotype the MHC of the Stokes skinkEgernia stokesiirevealed that 43 MHC I alleles were present in the offspring and two MHC I alleles were missing in its parents.This provides a useful reference for MHC genotyping in other taxa (Pearsonet al.,2017).In addition,clone sequencing and next-generation sequencing were used to type the MHC genes of 28 goat antelope,Rupicapra rupicapra,showing differences in MHC typing between the two methods in 25%of the individuals and again confirming the superior ability of next-generation sequencing to detect highly polymorphic genes(Stipoljevet al.,2020).
However,NGS methods can be prone to sequencing errors and false positives,i.e.,false “alleles”.To minimize the occurrence of false positives,sequencing depth and coverage can be increased,thus avoiding errors generated by sequencing,and additional bioinformatics methods can be developed to address these issues(Chenget al.,2022;Sebastianet al.,2018).Therefore,the use of NGS technology to obtain MHC gene sequences should take the corresponding sequencing depth into account.In the present study,NGS was used based on the amplicon principle,i.e.,using a high sequencing depth to exclude sequencing errors (the average amplicon reads were 62 533 base pairs) to ensure genotype accuracy.In addition,the deeper the sequencing depth,the higher the probability of detecting alleles,which could compensate for the limitations of difficult allele amplification in clone sequencing (differences in amplification efficiency between alleles)(Biedrzyckaet al.,2017).
4.3.Intraspecific and trans-species polymorphisms of MHC class I genesMHC genes form a highly polymorphic gene family with a higher rate of variable sites than other genes,and most animal MHC genes have genetic variability(Kohnet al.,2006).However,reptiles have low levels of genetic diversity in their MHC genes.The nucleotide diversity of different populations ofPodarcis erhardiiranged from 0 to 0.00564 (Hurstonet al.,2009).The nucleotide diversity of three populations in the southern Flinders Ranges ofE.stokesiiranged from 0.151 to 0.163(Pearsonet al.,2017).The π value of the MHC allele for 208Sphenodon punctatusfrom 12 island populations was 0.152±0.013.Compared to the above lizards,the π value ofE.multifasciatawas lower(0.101).The level of genetic diversity within a species can predict its evolutionary potential and viability(Guoet al.,2019).The higher the level of genetic diversity of the species,the smaller the impact of external environmental change,and the stronger the ability to survive(Paulset al.,2013).Multiple factors can influence the level of genetic diversity within a species,such as environmental variables,genetic drift within populations,bottleneck effects,inbreeding and life history traits,and diversity in a population.(Raposo Do Amaralet al.,2013).
The six species of lizards in this study are listed as threatened under three kinds of animal protection laws for the state,andS.indicusis assessed as a near-threatened species.Because of these classifications,and the vulnerability of these species,our sample sizes were small.The sample size may have affected the nucleotide diversity of the alleles to some extent.However,results generated from this study could help us gain a simple understanding of the level of genetic diversity in a given population,with implications for the conservation and management of particular populations.
Both the allele network and the phylogenetic tree showed that alleles were shared among the six lizard species in the family Scincidae,and they were not clustered by family in the phylogenetic tree.This indicated the existence of TSP in the MHC class I genes.This TSP could have arisen because the allelic lineage emerged before the species was formed and was retained after the species became established (Kleinet al.,1998).It could also be possible that the six species have overlapping ranges,with hybrid gradual infiltration between them(Guoet al.,2011),and that the evolutionary relationships exhibited by their MHC class I genes may be the result of hybridization.The phenomenon of TSP in MHC locus allele lineages has been reported in many vertebrates.For example,a phylogenetic tree constructed from exon 3 of the MHC class I genes of the oviparousE.brenchleyi,ovoviviparousE.multiocellataandE.przewalskiishowed that the MHC genes of the three species did not form their own genetic lineages according to species and did not show specific evolutionary lineage inheritance (Yuanet al.,2014).The phylogenetic tree showed that there was no interspecific mixing between the MHC class I alleles ofE.multiocellataand the three species of the generaEremias.It is therefore hypothesized that the divergence between the family Scincidae and Lacertidae occurred before the allelic lineage was formed.In addition,the phylogenetic relationships between the MHC I genes ofE.multiocellataand other lizards that are closely related were also studied,and alleles from each class did not fit neatly into several branches.This suggested that either the shared MHC class I genes ofE.multiocellatahad recently undergone too much gene duplication to exhibit homologous clustering of alleles,or that the alleles had not evolved independently after gene duplication(Winternitzet al.,2023).Some MHC class I alleles exhibit a greater degree of phylogenetic similarity between species than within species.This can be common for immune genes in general,which are subject to parasitemediated balancing selection and maintain allelic diversity over time(Těšický and Vinkler,2015).Convergent evolution,resulting from similar selective pressures,can also cause shared polymorphisms among independent evolutionary lineages(Krieneret al.,2000;Srithayakumaret al.,2012).
5.Conclusions
In most animals,the major histocompatibility complexes are polymorphic and polygenic,which makes genotyping problematic.This study demonstrated that when sequencing highly variable regions,such as the MHC,NGS technology performs better than clone sequencing.The level of genetic diversity of the MHC genes inE.multiocellatais low.In addition,the exon 3 sequences of the MHC gene of six species of lizards in the family Scincidae display TSP.This study contributes to our understanding of the MHC structure in lizards of the family Scincidae,and explores methods suitable for MHC genotyping in reptiles,while providing a reference for future studies on reptile genetic diversity.
Data availabilityThe raw data presented in the study have been uploaded on the SRA database from NCBI under the BioProject PRJNA962605.
AcknowledgementsWe would like to thank Wenjia SHEN for her lab assistance during the research.We thank Georgia WARD-FEAR and Zheng LI for their constructive comments on the revised version.This work was supported by grants from the National Natural Science Foundation of China(32171495 and 31971414),and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
杂志排行

Asian Herpetological Research的其它文章
- Oxygen Availability Affects Behavioural Thermoregulation of Turtle Embryos
- Sexual Dimorphism,Female Reproductive Characteristics and Embryonic Thermosensitivity in the Tonkin Forest Skink (Sphenomorphus tonkinensis)from Hainan,South China
- Evidence for Expensive Tissue Hypothesis in the Asian Common Toad(Duttaphrynus melanostictus)
- Redefinition of the Odorrana versabilis Group,with a New Species from China (Anura,Ranidae, Odorrana)
- A New Species of the Genus Nanorana Günther,1896 (Anura:Dicroglossidae) from Hengduan Mountains of China
- Response of Distribution Range Against Climate Change and Habitat Preference of Four National Protected Diploderma Species in Tibetan Plateau