柱后衍生离子色谱法测定土壤和沉积物中的六价铬
2023-12-26朱红霞周敬峰金淑聪胡佳欣
王 婷,朱红霞,周敬峰,姜 丽,金淑聪,胡佳欣
1.宝鸡市环境监测中心站,陕西 宝鸡 721000
2.中国环境监测总站,国家环境保护环境监测质量控制重点实验室,北京 100012
3.广西壮族自治区海洋环境监测中心站,广西 北海 536001
4.福建省生态环境监测中心,福建 福州 350000
5.昌吉州回族自治州环境监测站,新疆 昌吉 831100
铬(Cr)在自然界中广泛存在,主要以三价铬和六价铬存在[1]。 少量三价铬对人体有益[2],而六价铬毒性约为三价铬的100 倍[3],被认为是主要的人类致癌因素之一[4],可通过皮肤、胃肠道和呼吸系统被人体吸收,引起胃道及肝、肾功能损害等[5]。 六价铬在制革、纺织品生产、印染以及镀铬等行业有广泛应用,伴生的含铬废水若处理不当很容易渗入土壤[6]。 土壤对六价铬的吸附能力差,极易扩散迁移[7],因此,准确测定土壤中六价铬对于控制污染具有重要作用。
《土壤环境质量-建设用地土壤污染风险管控标准》(GB 36600—2018)将六价铬列为建设用地污染风险筛选和管制项目,但推荐的方法仅有碱溶液提取-火焰原子吸收分光光度法[8-9]。 现有对土壤中六价铬测定的报道主要基于特定技术提取,分光光度法[10-11]、火焰原子吸收法[12-14]和电感耦合等离子体发射光谱法[15-17]进行测定。 由于土壤基质复杂,相应提取液中干扰因素较多,应用分光光度法分析六价铬,土壤样品中的色度、还原性物质等易对测定结果产生干扰;应用原子吸收或电感耦合等离子体发射光谱等总铬的检测方法,则必须保证前处理提取出的目标物为六价铬,但目前对于提取液中铬形态的研究较少,提取液是否只有六价铬尚不明确,同时原子吸收测定时存在盐基沉积的问题[18]。 离子色谱法则弥补了这一缺陷[19],对于六价铬测定具有分离效果好、检出限低、进样量少等特点。 应用离子色谱法测定土壤中六价铬的研究相对较少,张涛等[2]选用超声辅助提取/离子色谱法测定了铬污染土壤中的六价铬,选用的检测器为电导检测器,相比紫外检测器测定六价铬灵敏度较低,且超声提取相比加热回流提取控温效果较差。
本研究选用氢氧化钠和碳酸钠强碱混合溶液作为提取剂,应用恒温搅拌回流提取,离子色谱分离,柱后衍生紫外可见检测器测定土壤六价铬,方法操作简便、灵敏度高、特异性强,是土壤中六价铬测定方法的有力补充。
1 实验部分
1.1 仪器、试剂与材料
1.1.1 试剂
氢氧化钠(优级纯,日本);碳酸钠(优级纯,德国);氯化钾(纯度≥99.8%,国药集团化学试剂有限公司);硝酸(55%,德国);氨水(25%,优级纯,国药集团化学试剂有限公司);碳酸氢钠(分析纯,国药集团化学试剂有限公司);浓硫酸(98%,优级纯,国药集团化学试剂有限公司);甲醇(色谱纯,德国);1,5-二苯基碳酰二肼(优级纯,国药集团化学试剂有限公司);六价铬标准储备液100 mg/L(生态环境部标准样品研究所);土壤六价铬质控样(ERA D113-921,美国)。
1.1.2 仪器和设备
柱后衍生离子色谱仪(紫外检测器,ELSpe-2型,广州谱临晟科技有限公司);加热回流装置(德润环保科技有限公司);水浴锅(BT47 型,yamato);超声波清洗器(KQ2200DE 型,昆山市超声仪器有限公司); 台式高速冷冻离心机(H2050R 型,长沙湘仪离心机仪器有限公司);电子天平(精确至0.1 mg,瑞士梅特勒公司);0.22 μm 亲水PTFE 针式滤器(上海安谱实验科技股份有限公司)。
1.1.3 条件实验用土壤样品
为提升方法普适性,选用7 种不同类型土壤或沉积物样品进行条件实验,编号为1 ~7 号,即:1 号GBW 07405(GSS-5)黄红壤,2 号GBW 07407(GSS-7)砖红壤,3 号GBW 07447(GSS-18)盐碱土, 4 号 GBW 07454 ( GSS-25 ) 黄土, 5 号GBW 07455(GSS-26)淮河沉积物,6 号承德市某地黄棕土,7 号承德市某地棕土。
1.2 实验方法
1.2.1 色谱条件
色谱柱为Prin-cen GF2108 六价铬快速分析柱4.1 mm×50 mm,5 μm;柱温为室温;流动相为0.1 mol/L HNO3+0.133 mol/L NH3·H2O,流速为1.2 m L/min;柱后衍生试剂为0.4 g 二苯碳酰二肼溶于50 m L 甲醇+50 m L 2 mol/L 硫酸定容到500 m L,流速为0.7 m L/m in;进样量为50 μL;检测器为紫外可见检测器;检测波长为540 nm。
1.2.2 样品前处理
筛分:参照《土壤环境监测技术规范》(HJ/T 166—2004),将土样风干后过孔径为0.150 mm 筛。
提取:称取2.5 g(精确至0.001 g)筛分后的试样于玻璃样品杯中,放入磁子,加入25.0 m L 提取剂(0.5 mol/L NaOH +0.28 mol/L Na2CO3),在样品杯外壁标记液面刻线,磁搅拌条件下120 ℃回流1 h,自然冷却后备用。 若冷却后提取液液面低于刻线,可用纯水补充至刻线后混匀待测。
净化:将样品杯中试样全量转入50 m L 塑料离心管,5 000 r/min 离心5 min。 将上清液用0.22 μm 的微孔滤膜过滤,弃去1 m L 初滤液,取适量稀释后上机待测。
2 结果与讨论
2.1 提取剂种类的选择
根据现有研究,选择4 种提取剂进行比对实验:第1 种,常用的氢氧化钠-碳酸钠溶液(0.5 mol/L NaOH +0.28 mol/L Na2CO3);第2 种,柱后衍生离子色谱法测定环境空气颗粒物中六价铬时使用碳酸氢钠溶液(20 mmol/L NaHCO3)[20];第3种,蔡晔等综合多种提取剂优选后的氢氧化钠-氯化钾溶液(0.5 mol/L NaOH +0.5 mol/L KCl)[21];第4 种,与实验室离子色谱测定六价铬条件一致的淋洗液(0.1 mol/L HNO3+0.133 mol/L NH3·H2O)。选用1.1.3 节中列出的7 种不同风干土壤或沉积物,向其中滴加适量六价铬标准溶液,待自然风干后制成7 种加标样品。 由于使用的实际样品为液体加标的土壤或沉积物样品,且风干后立即进行提取实验,其中的Cr(Ⅵ)与土壤或沉积物的结合并不牢固,故实验初期选择常用且操作便捷的超声提取,在不控温条件下使用4 种提取剂将7 种加标样品超声提取1 h,样品信息及加标回收率见表1。
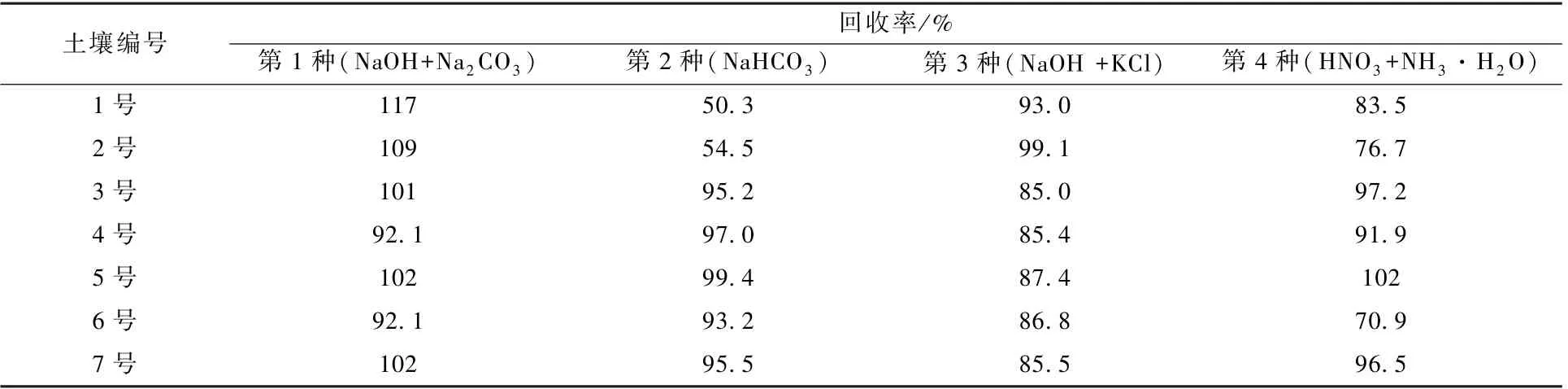
表1 不同提取剂回收率Table 1 Recovery of different extractants
由表1 可以看出:第2 种提取剂测定1 号、2号2 种土壤中六价铬时,回收率低至50%;第4 种提取剂测定2 号、6 号2 种土壤时,六价铬回收率低至70%。 第1 种(NaCO3+NaOH) 和第3 种(KCl+NaOH)2 种提取剂提取的7 种土壤的加标回收率为85%~120%,考虑到高浓度的KCl 易引起离子色谱柱饱和,故优选碳酸钠和氢氧化钠混合溶液作为提取剂。
2.2 提取方式的选择
常用可以满足混匀加热功能的提取方式包括超声提取、水浴振荡提取、恒温磁力搅拌回流提取。 参照《土壤和沉积物 六价铬的测定 碱溶液提取-火焰原子吸收分光光度法》(HJ 1082—2019)中对土壤六价铬的前处理条件,提取过程中至少需要将提取液加热至90 ℃,然而超声提取时水温波动较大,恒温的条件不易控制,且部分仪器水微沸时极易导致超声断开,故温度和超声的限制导致附着在土壤和沉积物晶格内的六价铬不易提取。 故仅比较了水浴振荡提取和恒温磁力搅拌回流装置对非加标样品的提取效果,在相同温度(90 ℃)和时间(60 m in)内提取并测定了7 种土壤中六价铬的本底含量,结果如图1 所示。
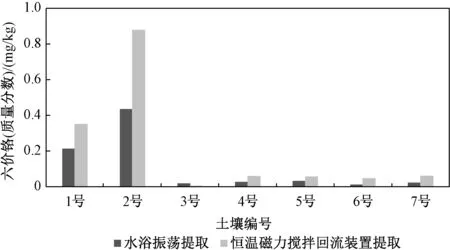
图1 2 种提取方式提取7 种土壤中六价铬Fig.1 Extraction of hexavalent chromium from 7 kinds of soil by 2 extraction methods
由图1 可以看出,恒温磁力搅拌回流提取出的六价铬含量明显高于水浴振荡提取,可能是实验室内的水浴振荡器对提取样的振动频率低于磁力搅拌,土壤中六价铬不能完全溶解在提取液中,故最终选用恒温磁力搅拌回流提取。
2.3 提取时间的选择
对2.1 节中制备的7 种加标样品进行实验,讨论提取时间。 参照现行方法标准或文献在10 ~60 min 内改变提取时间,比较测定回收率,结果见图2。
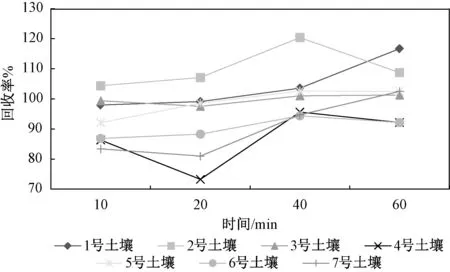
图2 提取时间对不同土壤加标回收率的影响Fig.2 Effect of extraction tim e on recovery rate of different soils
由图2 可以看出,提取时间超过40 min 时,7种土壤中添加六价铬标准溶液后的回收率均高于90%,考虑到土壤基质复杂,故仍参照HJ 1082—2019,加热回流提取60 m in。
2.4 提取温度的选择
应用上述7 种不同土壤和沉积物样品进行提取温度的讨论。 改变加热回流装置的底部加热温度,分别在常温(约25 ℃)、60 ℃、95 ℃、120 ℃、190 ℃条件下,比较各温度下样品本底中提取出的六价铬含量,结果见图3。 由图3 可以看出,提取温度对六价铬的提取效率影响较大,随温度升高,提取量逐渐增加。 在95 ℃时,实验所选的大部分样品中六价铬的测定值趋于稳定,但仍有2种样品的六价铬提取量未达到最大值。 在加热温度超过120 ℃后7 种样品均达到了最大提取量,故最终选取的提取温度为120 ℃。
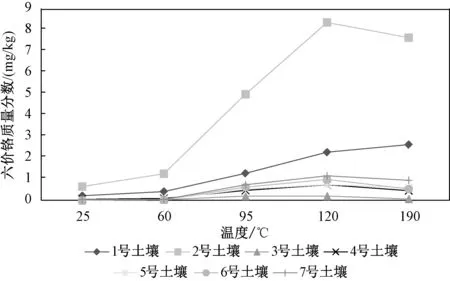
图3 不同的提取温度下不同土壤中六价铬的提取量Fig.3 The extraction amount of hexavalent chromium in different soil under different extraction temperature
2.5 提取剂用量的选择
通过改变提取剂体积和土壤质量比的方式选择最佳的提取剂用量,如图4 所示。 由图4 可以看出,固定土壤称样量和固定提取剂体积2 种方式得到的提取量较为一致,且提取剂体积和称样量的比大于10 ∶1 后,提取的土壤中的六价铬浓度趋于稳定,故最终选择提取剂体积和称样量比为 10 ∶1,根据本实验仪器设备规格,最终确定称取 2.5 g 土壤用25 m L 提取剂提取。
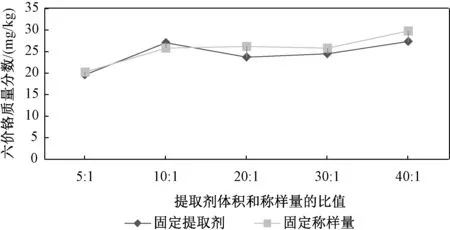
图4 提取剂体积和土壤的质量比对六价铬提取量的影响Fig.4 Effect of extractant volume and soil mass ratio on the extraction amount of hexavalent chromium
2.6 提取液的保存
选用3 号和6 号2 种土壤的提取液进行保存时间的影响实验,4 ℃冷藏14 d 内测定,结果如表2 所示。 从表2 可以看出,2 种土壤提取液冷藏时,14 d 内六价铬浓度变化不大,说明提取后可以在14 d 内完成测定。
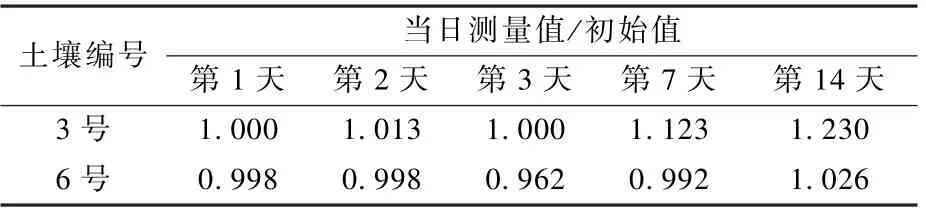
表2 提取剂保存时间对六价铬浓度的影响Table 2 Effect of storage time of extractant on concentration of hexavalent chromium
2.7 线性方程和检出限
用提取液配制0.0、2.0、5.0、10、30、50 μg/L的六价铬标准系列,应用离子色谱进行测定,标准曲线方程为y=60 852.736 8x+341.869 4,相关系数r=0.999 9,线性关系良好。
按照《环境监测分析方法标准制订技术导则》(HJ 168—2020)中空白实验未检出目标物的检出限测定要求,向25 m L 提取液中添加20 μL的1 mg/L 的标准溶液,配制成0.8 μg/L 的混合溶液,按照实际样品的测定步骤进行加热回流提取和测定,重复测定7 次,结果分别为0.762、0.780、0.815、0.754、0.907、0.880、0.849 μg/L,均值为0.821 μg/L,标准偏差为0.059 7 μg/L,该方法提取液中六价铬的检出限为0.2 μg/L。 以2.5 g 土壤称样量和25 m L 提取液体积计算,土壤中六价铬的方法检出限为0.002 mg/kg,远低于《土壤环境质量 建设用地土壤污染风险管控标准》(GB 36600—2018)中土壤六价铬的第一类用地筛选值(3.0 mg/kg)的限值要求,同时也低于李恒[17]用碱溶液提取-ICP 法测定土壤中的六价铬的方法检出限0.08 mg/kg;炼晓璐等[14]用碱消解-火焰原子吸收光谱法测定土壤中六价铬的方法检出限0.20 mg/kg;唐爱玲[18]用FAAS 和ICPMS 法测定污染土壤中六价铬,FAAS 法(火焰原子吸收分光光度法)测得的污染土壤中六价铬的方法检出限为0.44 mg/kg,ICP-MS 测得的检出限为0.003 mg/kg。 说明本方法适用于土壤中低含量六价铬的测定。
2.8 实际样品的测定
选择河北承德某地2 种实际土壤样品(编号为6 和7 的土壤)及编号为ERAD113-921[标准值为(25.5±1.5 )mg/kg]的六价铬标准土壤样品,按上述优化好的提取和离子色谱条件进行实际样品以及加标样品实验,平行测定6 次,具体结果如表3 所示,相对标准偏差为1.3%~3.7%,实际样品的回收率为93.6%~100%,标准样品的测定均值在给定范围内,对应离子色谱图如图5 和图6 所示,本方法适用于土壤中六价铬的测定。
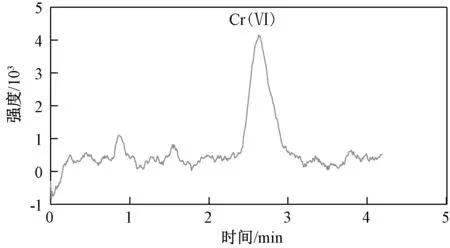
图5 样品6 的离子色谱图Fig.5 Ion chromatogram of sample 6
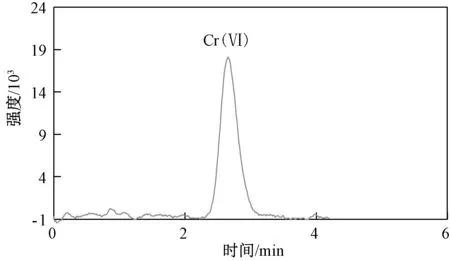
图6 样品7 的离子色谱图Fig.6 Ion chromatogram of sample 7
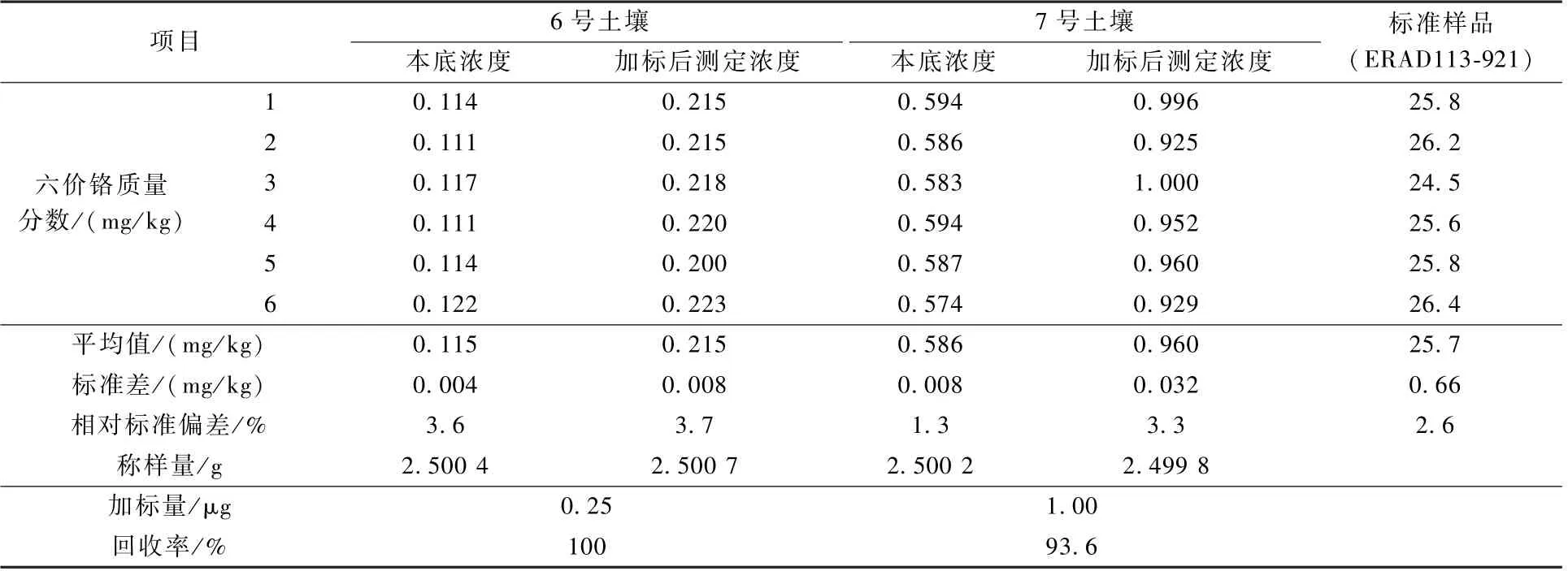
表3 实际样品和质控样测定结果Table 3 Test results of actual samples and quality control samples
3 结论
本文建立的加热回流提取、柱后衍生离子色谱法用于土壤中六价铬测定,检出限低至0.002 mg/kg,提取液损耗小,运行成本低,特异性强,准确度、精密度良好。 相比目前常用的原子吸收等土壤中铬(Ⅵ)测定方法,柱后衍生离子色谱法对于低含量土壤样品的测定更具优势,可满足土壤中低含量铬(Ⅵ)的测定需求。
