Ni、Fe杂质在LBE中的化学行为的深度势能分子动力学研究
2023-12-26梁汝婷聂长明柴之芳石伟群
梁汝婷,薄 涛,聂长明,张 蕾,柴之芳,石伟群
(1.南华大学 化学化工学院,湖南 衡阳 421001;2.中国科学院 宁波材料技术与工程研究所 先进能源材料实验室,浙江 宁波 315201;3.中国科学院 高能物理研究所 核能放射化学实验室,北京 100049)
当今核能领域正在大力探索第四代核电系统[1],其中铅冷快堆[2-3]是有望第一个实现商业化的堆型。液态铅铋共晶(LBE,44.5%Pb-55.5%Bi)由于其良好的中子特性、热物理性质和化学惰性[4],可用作铅冷快堆的冷却剂和散裂靶。LBE与传统结构材料(如奥氏体、铁素体和马氏体钢)的相容性是其在先进反应堆应用中面临的关键问题之一[5-7]。LBE对钢的腐蚀是由多种机制引起的,如金属元素在LBE中的溶解和扩散、在界面处形成金属间化合物以及液态金属沿晶界渗透。在高温和低氧浓度下,钢的主要金属成分Fe、Ni和Cr溶解在钢表面,并运输到LBE液体中,当金属杂质浓度在LBE中达到饱和时,溶解过程停止,否则会持续腐蚀[8-10]。金属杂质的主要运输机制为原子向钢表面静止的流体层和相邻的流动层扩散,金属杂质的扩散速率取决于其在LBE中的浓度梯度和扩散系数。然而,目前对液态LBE中杂质原子扩散现象的认识仍然有限,在高温下测量液态LBE中金属杂质的扩散系数尤其困难,目前这方面的研究还很少。因此,本文拟采用理论模拟的方法研究LBE中金属杂质扩散性质。
凝聚态系统的分子动力学(MD)模拟越来越多地应用于工程领域,它可以提供难以获得的实验数据和对相关物理化学性质机理的基本见解[11]。模拟方法的选择取决于系统的特性和期望的效果。从头算分子动力学(AIMD)[12]用于模拟液态金属是完全可行的,该方法结合密度泛函理论(DFT)[13-14]和Car-Parrinello(CP)方法[15],从量子力学电子密度出发,对原子核上的力进行“动态”计算,可准确地描述电子与分子之间的结构、性质和相互作用。AIMD能提供重要的结构和电子信息[16](如对分布函数、态的电子密度、成键和电荷转移),Gil等[17]研究了元素周期表第二排杂质原子在LBE中的微观结构和电荷转移;Ding等[18]研究了杂质原子在LBE中的能量关系和键合强度。然而,随着原子数、模型尺寸和模拟时间的增加,计算成本呈指数增长,而扩散是一种相对缓慢的过程,需要1 ns或更长时间才能清楚地分辨出来,这限制了AIMD的应用。此外,由于系统尺寸的限制,在100个Pb和Bi原子中只允许引入1个杂质原子,在这种情况下,原子的均方根位移(决定其扩散系数)的统计数据将太过嘈杂而无法使用。使用传统经验势的经典MD,在模拟体系和效率上都更具有优势,它可以进行上万个原子空间尺度以及ns级时间尺度的模拟,但传统经验势需要获得大量的原子间参数,耗时费力,且传统经验势的适用体系范围小,在原始参数范围之外应用时需进行仔细测试,准确性难以保证。Zhou等[19]使用嵌入原子势(EAM)力场研究了Pb和Bi在钢表面的渗透深度;Gao等[20]计算了LBE的热物理学性质,但其结果与实验值有一定差距,得到的径向分布函数(RDF)也达不到AIMD的精度。近年来,基于机器学习(ML)的原子间势能得到广泛应用,Zhang等[11-21]提出的深度势能(DP)方法被证明是一种精确、高效的大规模长时间MD模拟方法,可以成功地解决上述精度与效率的难题。该方法从AIMD计算中选择数据集进行训练,通过构建深度神经网络(DNN)[22],将原子的局部环境自动编码为对称保持描述符,再将描述符映射到原子势能。经过充分训练的DP可以达到DFT的精度。同时,DP具有传统经验势的计算效率,可以计算上万原子体系和数百纳秒的时间尺度。目前深度势能分子动力学(DPMD)已应用于众多领域,并取得了优异的成果[23-27]。
本文拟基于DPMD方法,使用深度势能生成软件DP-GEN和DeePMD-Kit对液态LBE、Ni和Fe分别溶解于LBE中(LBE-Ni和LBE-Fe)的3个体系的DP模型进行训练,并与DFT结果进行对比,以验证DP模型的精度,最后用MD软件LAMMPS计算3个体系的密度、热容、黏度和杂质原子Ni、Fe在LBE中的RDF、配位数和扩散系数。
1 计算方法
1.1 深度神经网络势能训练
使用DeePMD-kit软件[28]进行DP训练,从训练集中的原子坐标信息(包括原子间相对径向坐标和角坐标)出发,通过DNN构建复杂材料体系的势能面。某一体系的势能E由i类原子中每类原子的能量Ei的加和组成,即:
(1)
(2)
其中:Δε、ΔFi分别为DP模型预测结果与训练数据集(DFT计算得到)之间的能量和力的差值;pε、pf和pξ分别为能量、力和维里张量的权重系数。本文在训练过程中并未考虑维里张量,p从0.02增加到1.00,pf从1 000减少到1。LBE、LBE-Ni、LBE-Fe的DP模型分别用2 000 000、1 000 000、3 000 000步进行训练。
1.2 主动学习生成训练集
(3)
其中,〈·〉 表示模型预测的平均值。
其次,空空如也的木桶是多么沉重!因为他装载的不只是煤炭,更是鲜活的肉身,这活着的该死的肉身实在太沉重,他既有尊严感却又偏偏需要物资的侍候,想象一下骑桶者每次提着空桶前去借煤时的惶恐不安且借而不得的绝望与屈辱,他该有多么痛恨这肉身的沉重!为什么肉身就不能轻盈起来呢?——必须骑着木桶前去,如此就好像把沉重的生活骑在了胯下;必须骑着木桶前去,如此肉身就轻盈起来;轻盈的肉身,把沉重的生活骑在胯下的肉身,是活着的根本目的。——木桶必须飞起来。显然,这是木桶飞起来的生存论依据,他超越于地球引力之上,是合乎情理的心理现实。

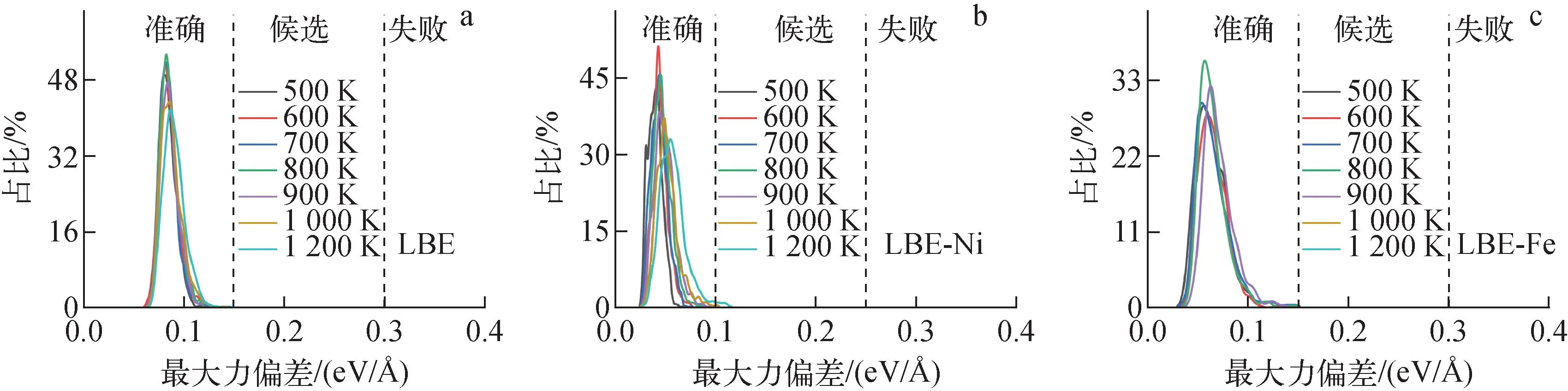
图1 DP模型的最大力偏差Fig.1 Maximum deviation of DP model prediction of force
由图1可见,在探索数据集的最后一步,LBE、LBE-Ni和LBE-Fe三个体系的原子最大力偏差集中在小于0.1 eV/Å的范围内,说明收集到的数据集足够丰富,LBE、LBE-Ni和LBE-Fe体系分别包含2 000、2 900、1 500个训练数据集。最终,使用DPGEN探索到的训练数据集对3个体系分别进行长步数训练。
1.3 DFT计算
DP-GEN训练的初始训练数据集通过AIMD模拟获取,标记步骤中得到的候选结构需要进行DFT自洽计算。本文使用第一性原理软件包VASP[33-34]进行所有DFT计算,用投影缀加平面波(PAW)[35-36]方法描述电子与离子的相互作用,用Perdew-Burke-Ernzerhof (PBE)[37]泛函描述电子间交换关联作用,平面波截断能为500 eV,K点设置为1×1×1。对于所有AIMD计算,采用NVT系综和Nosé-Hoover恒温器,时间步长设置为1 fs。初始结构使用Packmol程序[38]生成,对于LBE体系,初始结构由3种原子比例(Pb∶Bi=30∶70、45∶55、70∶30)和3个原子间距(2.0、2.1、2.2 Å)的9个结构通过DFT结构优化后得到,9个初始结构在700 K下模拟0.1 ps得到900个初始数据集;同时,训练了加入范德华校正后的LBE(DFT-D3)体系和LBE(optB86b-vdW)体系的两种DP力场,LBE(DFT-D3)体系的初始结构由3种原子比例(Pb∶Bi=30∶70、45∶55、70∶30)和3个原子间距(2.0、2.1、2.2 Å)的9个结构加入DFT-D3校正进行结构优化后得到,9个初始结构在600、800、1 000、1 200、1 400 K下模拟0.005 ps得到225个初始数据集;LBE(optB86b-vdW)体系的初始结构由3种原子比例(Pb∶Bi=30∶70、45∶55、70∶30),3个原子间距(2.0、2.1、2.2 Å)的9个结构加入optB86b-vdW校正进行结构优化后得到,初始数据集由LBE(DFT-D3)体系的验证集加入optB86b-vdW校正后得到。对于LBE-Ni体系,初始结构由3种原子比例(Pb∶Bi∶Ni=45∶55∶1、45∶55∶3、45∶55∶5),4个原子间距(1.9、2.0、2.1、2.2 Å)的12个结构进行DFT结构优化后得到,12个初始结构分别在600、700、800、900 K下模拟0.01 ps,得到480个初始训练数据集;对于LBE-Fe体系,初始结构由3种原子比例(Pb∶Bi∶Fe=30∶70∶5、50∶50∶5、70∶30∶5),原子间距为2.0 Å的3个结构进行DFT结构优化后得到,3个初始结构在700 K下,模拟0.1 ps,共300个初始训练数据集。
1.4 MD模拟设置
使用MD软件LAMMPS[39]进行DPMD模拟,所有模拟都采用周期性边界条件来消除边界效应。用Verlet算法求解牛顿运动方程的时间步长为1 fs。在使用DPGEN探索数据集步骤中,先在NPT系综下迭代几次后再用NVT系综,可以丰富数据集,温度范围设为400~1 200 K,压力范围设为-105~2×108Pa。训练结束后用DP模型进行DPMD模拟,模拟方法如下:各体系首先在NPT系综下平衡60 ps,得到平衡后的结构;然后采用NPT系综模拟0.3 ns计算热容和密度;采用NVT系综模拟2 ns,计算RDF、CN、扩散系数和黏度。用于DPMD模拟的初始体系由packmol建模,其中,LBE体系包含3 584个Pb原子和4 416个Bi原子;LBE-Ni体系包含2 880个Pb原子、3 520个Bi原子和64个Ni原子;LBE-Fe体系包含3 584个Pb原子、4 416个Bi原子和20个Fe原子。
2 结果讨论
2.1 DP训练和验证
对DPGEN探索到的训练数据集进行长步数训练后的均方根误差(RMSE)结果如图2所示,可见在经过所设定步数的训练后,RMSE均趋向于收敛,DP模型得到了充分训练。训练结束后,通过对验证数据集进行测试来检验DP模型的准确性,验证数据集是在DPGEN探索收敛后,由新的初始结构进行下一次DPGEN迭代生成,LBE体系的验证数据集由若干Pb45Bi55、Pb30Bi70和Pb70Bi30组成,共300个;LBE-Ni体系的验证集由Pb45Bi55Ni1、Pb45Bi55Ni3和Pb45Bi55Ni5组成,共241个;LBE-Fe体系的验证集由300个Pb30Bi70Fe5、Pb45Bi55Fe5和Pb70Bi30Fe5数据组成。验证数据集的DP与DFT计算的能量和受力的对比如图3所示,可见3个体系的DP与DFT能量差的绝对值都小于0.01 eV/atom,力的差值在0.3 eV/Å以下,说明训练的DP力场可以准确预测体系在较宽温度和压强范围的能量和原子受力。

a,d——LBE体系;b,e——LBE-Ni体系;c,f——LBE-Fe体系图2 DP模型训练过程中训练数据集、验证数据集、能量和受力的均方根误差Fig.2 Training errors in training processes of DP model

a~c——LBE体系;d~h——LBE-Ni体系图4 773 K下DPMD与AIMD计算RDF的结果对比Fig.4 RDF results from AIMD and DPMD simulations at 773 K
为进一步验证DP的准确性,对比了DPMD和AIMD计算3个体系RDF的结果,如图4所示。可以看出,虽然由于AIMD模拟时LBE-Ni体系中Ni原子数量的限制,Bi-Ni和Pb-Ni的RDF较粗糙,整体上DPMD与AIMD预测的3个体系的RDF的结果吻合良好。
2.2 微观结构

图5 不同温度下的RDF和773 K下Ni、Fe原子的CNFig.5 RDF results from DPMD simulations at different temperatures and coordination numbers of Ni and Fe and at 773 K
2.3 热物理性质
1) 热容和密度
LBE热容的实验数据很少[40],且差异大,在液态金属手册(Liquid-Metals Handbook)[41]中Lyon推荐LBE在417~673 K温度范围内为定值146.5 J/(kg·K),在后续的出版物[42]中Kutateladze将温度范围扩大至403~973 K。Hultgren等[43]则认为LBE的热容随温度的升高而下降,这也与二元金属的柯普定律相符,LBE手册[10]根据这些数据给出了400~1 100 K内LBE比定压热容(cp,LBE,J/(kg·K))的拟合曲线:
cp(LBE)=164.8-3.94×10-2T+1.25×
10-5T2+1.25×10-5-4.65×10-5T-2
(4)
目前关于LBE热容的计算数据有限,本文的DPMD结果可以提供参考值,采用DPMD计算热容的公式如下:
(5)
其中,H、T、ρ、V分别为焓、温度、密度、体积,密度和焓取NPT系综模拟的0.3 ns内的平均值进行计算。
比定压热容的DPMD计算值与文献结果的对比如图6a所示。由图6a可见,本文采用DPMD计算得到的LBE比定压热容在673~973 K温度内,从140.19 J/(kg·K)下降到133.66 J/(kg·K),与LBE手册推荐值的相对误差为2.61%~3.35%,可见DPMD计算的结果非常准确。
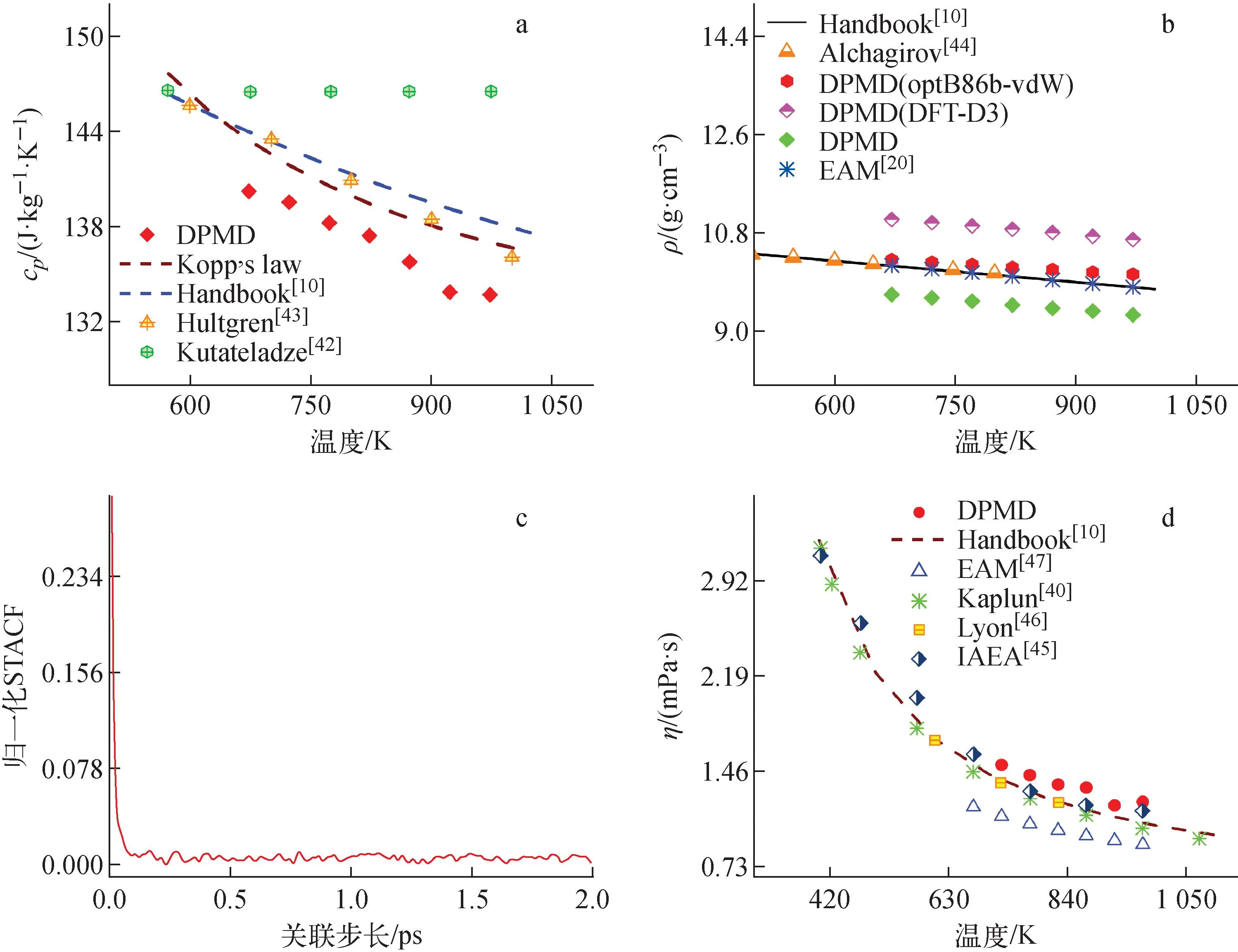
图6 LBE热物理性质DPMD计算值与文献值的对比以及LBE体系应力张量自相关函数随时间的变化Fig.6 Comparison between values of LBE thermophysical properties by DPMD and in literature and normalized stress tensor ACF at 773 K
LBE手册对400~1 273 K内的密度推荐值为ρLBE(kg/m3)=11 065-1.293T。
本文分别用未加vdW校正(图6b中DPMD)、加入optB86b-vdW和DFT-D3校正的3个DP力场计算了LBE的密度,并与文献数据进行对比,结果示于图6b。由图6b可见,DPMD的计算结果较LBE手册推荐值[10]略小,相对误差在5.09%~5.18%内;而optB86b-vdW校正的计算结果与LBE手册推荐值非常接近,相对误差在1.18%~2.36%内;DFT-D3校正的计算结果较LBE手册推荐值大,相对误差在8.42%~8.90%内。可以看出,加入optB86b-vdW校正后能得到更准确的密度值,这是由于PBE泛函未考虑弱相互作用,原子间距离不够紧密,体积偏大,导致密度偏低,引入合适的vdW校正可以解决这一问题。optB86b-vdW校正后计算得到的密度在673~973 K内从10.31 g/cm3下降到10.04 g/cm3,与实验值[44]及Gao等[20]使用EAM力场的计算值符合较好。
2) 黏度
黏度是影响LBE流动和杂质原子扩散的重要因素,其计算公式为:
(6)
其中:kB为玻尔兹曼常数;Sxy为应力张量的分量。
本文使用Green-Kubo方法,通过应力张量自相关函数(ACF)与时间的积分计算LBE的黏度。在Green-Kubo方法中,计算ACF的时间应该足够长,以确保归一化的应力张量ACF可以衰减为0,由图6c可看出,2 ps已经适用。基于此计算了723~973 K温度下LBE的黏度,结果如图6 d所示。由图6d可见,LBE黏度随温度的升高而下降,与实验值[40,45-46]趋势一致,DPMD结果与LBE手册拟合值的相对误差为7.22%~14.3%,Gao等[47]用EAM力场的计算值与手册推荐值的相对误差为16.03%~20.41%,相比之下,DPMD的结果更贴近实验值,这也验证了DP模型的准确性。
3) 扩散系数
扩散系数(D)由均方根位移(MSD)随时间的变化关系推导得出:
MSD=〈|δri(t)|2〉=
(7)
(8)
其中,δri为i类原子在时间t内的位移。
Ni、Fe原子不同温度下的MSD曲线如图7a、b所示,可见其MSD曲线并不平滑,这是由于杂质原子数量较少,其中Ni原子在LBE中的扩散速度较Fe原子更快。根据式(9)计算了Ni、Fe原子的扩散系数,结果如图7c、d所示。实验上关于杂质原子的扩散系数数据有限,Gao等[48]采用毛细管法测定了Ni原子在823、873、923 K LBE中扩散3.5 h和7 h的扩散系数,但由于溶解过程中Ni粉末与液态LBE边界层Ni、Pb和Bi固体的影响,其结果与Stoke-Einstein方程所得结果有所偏离,而本文的DPMD结果(图7c)则与Stoke-Einstein方程吻合较好,且与EAM结果[47]相比,DPMD预测的准确性更好。图7d为Fe原子的扩散系数,与EAM的预测结果相比,本文的DPMD结果更贴近手册值。根据DPMD计算结果拟合出Ni和Fe在673~973 K下的扩散系数(D,cm2/s)公式如下:
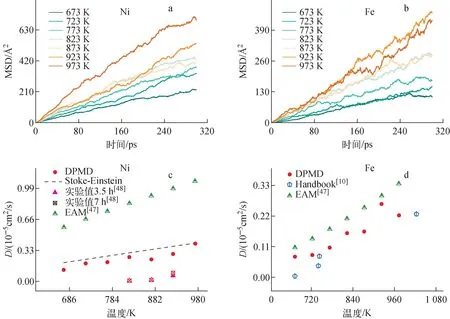
图7 不同温度下Ni、Fe原子在LBE中的圴方根位移与扩散系数Fig.7 MSDs and diffusion coefficient versus time for Fe atoms and Ni atoms diffusing in LBE at different temperatures
DNi=3.58×10-4exp(-1.85×104/RT)
(9)
DFe=4.65×10-4exp(-2.35×104/RT)
(10)
3 结论
本文首先训练了LEB、LBE-Ni和LBE-Fe的DP模型,然后用它们预测了微观结构和热物理性质。结果表明,本文训练的3个DP模型能准确描述LEB、LBE-Ni和LBE-Fe体系的能量和受力。3个DP模型计算得到的LEB、LBE-Ni和LBE-Fe体系的能量和受力与DFT计算结果对比,误差分别在0.01 eV/atom和0.3 eV/Å以下。3个DP模型用于计算RDF能重复AIMD的结果。不同温度下RDF的计算结果显示,温度升高,原子间的作用力减小,杂质原子与Bi原子均有更强的结合能力。根据RDF峰位置得到了各原子间的距离,计算了第一配位层的CN,两种杂质原子的配位情况相同,Pb-Ni、Pb-Fe的CN都是2和3,Bi-Ni、Bi-Fe的CN都是4。DPMD计算得到的LBE热容随温度升高而减小,计算值与手册值最大相对误差为3.35%。计算LBE密度时,加入optB86b-vdW校正后能得到更准确的结果,计算结果与实验值的相对误差在1.18%~2.36%内。723~973 K下,黏度在1.95~1.22 Pa·s范围内,且随温度的升高而减小,略高于实验值。由MSD结果计算得到了杂质原子的扩散系数,Ni、Fe原子的扩散系数随温度的升高而增大,Ni原子在LBE中的扩散速度大于Fe原子。以上结果显示,DP模型无论预测黏度还是扩散系数都较EAM有更好的准确性。
基于上述模拟结果,采用训练后的DP模型可以深入了解杂质原子在LBE中的微观结构,丰富LBE的热物理性质数据,有利于LBE在核能系统的应用。
